Jingjing Yang, Zijian Li, Ruiyi Ma, Shijie Xie, Dan Wang, Rong Quan, Jue Liu, Jiangwei Song
{"title":"塞内卡谷病毒3C蛋白酶靶向TRIM32裂解,拮抗其抗病毒作用。","authors":"Jingjing Yang, Zijian Li, Ruiyi Ma, Shijie Xie, Dan Wang, Rong Quan, Jue Liu, Jiangwei Song","doi":"10.1128/jvi.00590-25","DOIUrl":null,"url":null,"abstract":"<p><p>The Seneca Valley virus (SVV), a newly emerged virus within the <i>Picornaviridae</i> family, causes porcine idiopathic vesicular disease, imposing substantial economic costs to the global pork industry. However, the molecular mechanisms underlying its pathogenesis remain poorly understood. In this study, we identified TRIM32 as a novel antiviral factor against SVV and found that SVV infection led to the cleavage and degradation of TRIM32. Knocking down TRIM32 increased viral replication, while overexpressing it reduced viral titers. Further study showed that TRIM32 restricts SVV replication by selectively targeting viral VP3 protein for ubiquitination and proteasomal degradation. To counteract this, SVV 3C protease (3Cpro) targets TRIM32 for cleavage at E332. This cleavage renders TRIM32 unable to inhibit SVV replication or block the degradation of VP3. Additionally, the cleaved TRIM32 products weaken E3 ubiquitin ligase activity and reduce activation of the type I interferon (IFN) pathway. Taken together, our results indicate that SVV 3C<sup>pro</sup>-mediated cleavage of TRIM32 impairs its function in the ubiquitination and degradation of viral VP3 and type I IFN signaling. These findings offer novel insights into the strategies viruses use to evade the host's antiviral immune responses, thereby contributing to efficient viral replication.</p><p><strong>Importance: </strong>Seneca Valley virus (SVV) is an emerging pathogen that causes vesicular diseases in pigs, posing a significant threat to the global swine industry. Tripartite motif-containing protein (TRIM) family members are recognized as intrinsic antiviral effectors that provide a frontline shield against viruses prior to the transcription of interferon (IFN) and interferon-stimulated genes (ISGs). In this study, we uncovered the antiviral mechanism, which promotes the K48-linked ubiquitination of viral VP3 protein, leading to the degradation of VP3 via the proteasome pathway. SVV 3C<sup>pro</sup> abolished the antiviral effects of TRIM32 by inducing its cleavage. The cleaved TRIM32 fragment attenuates its E3 ubiquitin ligase activity and weakens the activation of IFN signaling. Our results reveal a potential mechanism of viral immune evasion, which is crucial for understanding how SVV has evolved a novel strategy to evade the intrinsic cellular restrictions against viral infection.</p>","PeriodicalId":17583,"journal":{"name":"Journal of Virology","volume":" ","pages":"e0059025"},"PeriodicalIF":3.8000,"publicationDate":"2025-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12455967/pdf/","citationCount":"0","resultStr":"{\"title\":\"Seneca Valley virus 3C protease targets TRIM32 for cleavage to antagonize its antiviral effects.\",\"authors\":\"Jingjing Yang, Zijian Li, Ruiyi Ma, Shijie Xie, Dan Wang, Rong Quan, Jue Liu, Jiangwei Song\",\"doi\":\"10.1128/jvi.00590-25\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The Seneca Valley virus (SVV), a newly emerged virus within the <i>Picornaviridae</i> family, causes porcine idiopathic vesicular disease, imposing substantial economic costs to the global pork industry. However, the molecular mechanisms underlying its pathogenesis remain poorly understood. In this study, we identified TRIM32 as a novel antiviral factor against SVV and found that SVV infection led to the cleavage and degradation of TRIM32. Knocking down TRIM32 increased viral replication, while overexpressing it reduced viral titers. Further study showed that TRIM32 restricts SVV replication by selectively targeting viral VP3 protein for ubiquitination and proteasomal degradation. To counteract this, SVV 3C protease (3Cpro) targets TRIM32 for cleavage at E332. This cleavage renders TRIM32 unable to inhibit SVV replication or block the degradation of VP3. Additionally, the cleaved TRIM32 products weaken E3 ubiquitin ligase activity and reduce activation of the type I interferon (IFN) pathway. Taken together, our results indicate that SVV 3C<sup>pro</sup>-mediated cleavage of TRIM32 impairs its function in the ubiquitination and degradation of viral VP3 and type I IFN signaling. These findings offer novel insights into the strategies viruses use to evade the host's antiviral immune responses, thereby contributing to efficient viral replication.</p><p><strong>Importance: </strong>Seneca Valley virus (SVV) is an emerging pathogen that causes vesicular diseases in pigs, posing a significant threat to the global swine industry. Tripartite motif-containing protein (TRIM) family members are recognized as intrinsic antiviral effectors that provide a frontline shield against viruses prior to the transcription of interferon (IFN) and interferon-stimulated genes (ISGs). In this study, we uncovered the antiviral mechanism, which promotes the K48-linked ubiquitination of viral VP3 protein, leading to the degradation of VP3 via the proteasome pathway. SVV 3C<sup>pro</sup> abolished the antiviral effects of TRIM32 by inducing its cleavage. The cleaved TRIM32 fragment attenuates its E3 ubiquitin ligase activity and weakens the activation of IFN signaling. Our results reveal a potential mechanism of viral immune evasion, which is crucial for understanding how SVV has evolved a novel strategy to evade the intrinsic cellular restrictions against viral infection.</p>\",\"PeriodicalId\":17583,\"journal\":{\"name\":\"Journal of Virology\",\"volume\":\" \",\"pages\":\"e0059025\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2025-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12455967/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Virology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1128/jvi.00590-25\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Virology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1128/jvi.00590-25","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/12 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
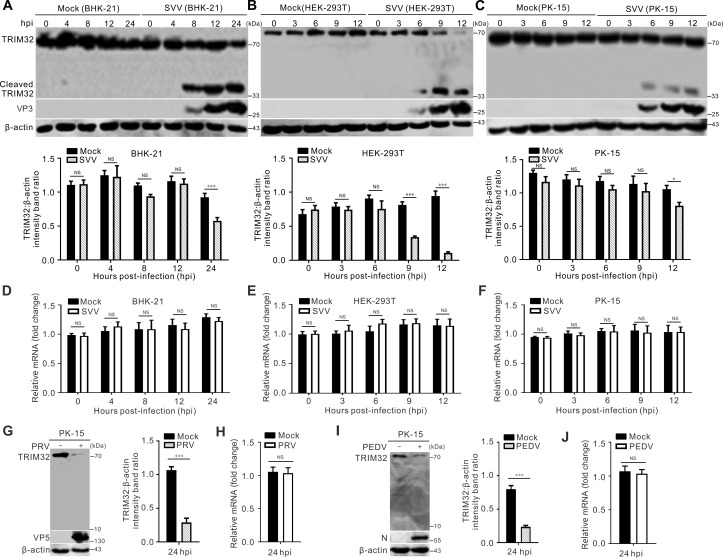
塞内卡谷病毒(SVV)是小核糖核酸病毒科中一种新出现的病毒,可引起猪特发性水疱病,给全球猪肉产业造成巨大的经济损失。然而,其发病机制的分子机制仍然知之甚少。在本研究中,我们发现TRIM32是一种针对SVV的新型抗病毒因子,并发现SVV感染导致TRIM32的裂解和降解。敲除TRIM32可增加病毒复制,而过表达TRIM32可降低病毒滴度。进一步研究表明,TRIM32通过选择性靶向病毒VP3蛋白泛素化和蛋白酶体降解来限制SVV的复制。为了抵消这一点,SVV 3C蛋白酶(3Cpro)靶向TRIM32在E332处进行切割。这种切割使得TRIM32无法抑制SVV的复制或阻止VP3的降解。此外,被切割的TRIM32产物削弱E3泛素连接酶活性,降低I型干扰素(IFN)途径的激活。综上所述,我们的研究结果表明,SVV 3cpro介导的TRIM32切割损害了其在病毒VP3和I型IFN信号的泛素化和降解中的功能。这些发现为病毒用来逃避宿主抗病毒免疫反应的策略提供了新的见解,从而有助于有效的病毒复制。重要性:塞内卡谷病毒(SVV)是一种引起猪水疱性疾病的新兴病原体,对全球养猪业构成重大威胁。Tripartite motifi -containing protein (TRIM)家族成员被认为是内在的抗病毒效应物,在干扰素(IFN)和干扰素刺激基因(ISGs)转录之前为病毒提供一线屏障。在这项研究中,我们揭示了抗病毒机制,该机制促进了病毒VP3蛋白的k48连锁泛素化,通过蛋白酶体途径导致VP3的降解。SVV 3Cpro通过诱导TRIM32的切割来消除其抗病毒作用。被切割的TRIM32片段降低了其E3泛素连接酶活性,减弱了IFN信号的激活。我们的研究结果揭示了病毒免疫逃避的潜在机制,这对于理解SVV如何进化出一种新的策略来逃避针对病毒感染的内在细胞限制至关重要。
Seneca Valley virus 3C protease targets TRIM32 for cleavage to antagonize its antiviral effects.
The Seneca Valley virus (SVV), a newly emerged virus within the Picornaviridae family, causes porcine idiopathic vesicular disease, imposing substantial economic costs to the global pork industry. However, the molecular mechanisms underlying its pathogenesis remain poorly understood. In this study, we identified TRIM32 as a novel antiviral factor against SVV and found that SVV infection led to the cleavage and degradation of TRIM32. Knocking down TRIM32 increased viral replication, while overexpressing it reduced viral titers. Further study showed that TRIM32 restricts SVV replication by selectively targeting viral VP3 protein for ubiquitination and proteasomal degradation. To counteract this, SVV 3C protease (3Cpro) targets TRIM32 for cleavage at E332. This cleavage renders TRIM32 unable to inhibit SVV replication or block the degradation of VP3. Additionally, the cleaved TRIM32 products weaken E3 ubiquitin ligase activity and reduce activation of the type I interferon (IFN) pathway. Taken together, our results indicate that SVV 3Cpro-mediated cleavage of TRIM32 impairs its function in the ubiquitination and degradation of viral VP3 and type I IFN signaling. These findings offer novel insights into the strategies viruses use to evade the host's antiviral immune responses, thereby contributing to efficient viral replication.
Importance: Seneca Valley virus (SVV) is an emerging pathogen that causes vesicular diseases in pigs, posing a significant threat to the global swine industry. Tripartite motif-containing protein (TRIM) family members are recognized as intrinsic antiviral effectors that provide a frontline shield against viruses prior to the transcription of interferon (IFN) and interferon-stimulated genes (ISGs). In this study, we uncovered the antiviral mechanism, which promotes the K48-linked ubiquitination of viral VP3 protein, leading to the degradation of VP3 via the proteasome pathway. SVV 3Cpro abolished the antiviral effects of TRIM32 by inducing its cleavage. The cleaved TRIM32 fragment attenuates its E3 ubiquitin ligase activity and weakens the activation of IFN signaling. Our results reveal a potential mechanism of viral immune evasion, which is crucial for understanding how SVV has evolved a novel strategy to evade the intrinsic cellular restrictions against viral infection.
期刊介绍:
Journal of Virology (JVI) explores the nature of the viruses of animals, archaea, bacteria, fungi, plants, and protozoa. We welcome papers on virion structure and assembly, viral genome replication and regulation of gene expression, genetic diversity and evolution, virus-cell interactions, cellular responses to infection, transformation and oncogenesis, gene delivery, viral pathogenesis and immunity, and vaccines and antiviral agents.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


