Hannah Gillespie, Yi Shiau Ng, Katrina M Wood, Sila Hopton, Charlotte L Alston, Emma L Blakely, Nick Thompson, Robert W Taylor, Andrew C Browning, Robert McFarland, John A Sayer
{"title":"m.3243A>T变异所致线粒体疾病的严重临床表现:早发、多器官受累、早死1例","authors":"Hannah Gillespie, Yi Shiau Ng, Katrina M Wood, Sila Hopton, Charlotte L Alston, Emma L Blakely, Nick Thompson, Robert W Taylor, Andrew C Browning, Robert McFarland, John A Sayer","doi":"10.1007/s44162-025-00110-0","DOIUrl":null,"url":null,"abstract":"<p><p>The spectrum of disease associated with pathogenic mitochondrial DNA (mtDNA) variants is wide. Most often, heteroplasmic mitochondrial DNA disease is the result of an adenine to guanine transition at position 3243 of mtDNA (m.3243A > G) in the <i>MT-TL1</i> gene encoding tRNA<sup>Leu(UUR)</sup>. Here, we present a case of a patient with a rarer m.3243A > T variant whose phenotype was severe and included delayed growth, developmental delay, myoclonic jerks and tonic-clonic seizures, progressive myopathy, cerebellar ataxia, severe malnutrition due to intestinal dysmotility despite naso-jejunal feeding requiring total parenteral nutrition, bilateral sensorineural hearing loss, and visual impairment, including bilateral cataracts requiring treatment and pigmentary retinopathy. At age 18 years, he developed severe nephrotic syndrome secondary to a membranoproliferative pattern of glomerular injury, which was resistant to treatment and led to premature death.</p>","PeriodicalId":73925,"journal":{"name":"Journal of rare diseases (Berlin, Germany)","volume":"4 1","pages":"47"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12334418/pdf/","citationCount":"0","resultStr":"{\"title\":\"Severe clinical manifestation of mitochondrial disease due to the m.3243A>T variant: a case report of early-onset, multi-organ involvement and premature death.\",\"authors\":\"Hannah Gillespie, Yi Shiau Ng, Katrina M Wood, Sila Hopton, Charlotte L Alston, Emma L Blakely, Nick Thompson, Robert W Taylor, Andrew C Browning, Robert McFarland, John A Sayer\",\"doi\":\"10.1007/s44162-025-00110-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The spectrum of disease associated with pathogenic mitochondrial DNA (mtDNA) variants is wide. Most often, heteroplasmic mitochondrial DNA disease is the result of an adenine to guanine transition at position 3243 of mtDNA (m.3243A > G) in the <i>MT-TL1</i> gene encoding tRNA<sup>Leu(UUR)</sup>. Here, we present a case of a patient with a rarer m.3243A > T variant whose phenotype was severe and included delayed growth, developmental delay, myoclonic jerks and tonic-clonic seizures, progressive myopathy, cerebellar ataxia, severe malnutrition due to intestinal dysmotility despite naso-jejunal feeding requiring total parenteral nutrition, bilateral sensorineural hearing loss, and visual impairment, including bilateral cataracts requiring treatment and pigmentary retinopathy. At age 18 years, he developed severe nephrotic syndrome secondary to a membranoproliferative pattern of glomerular injury, which was resistant to treatment and led to premature death.</p>\",\"PeriodicalId\":73925,\"journal\":{\"name\":\"Journal of rare diseases (Berlin, Germany)\",\"volume\":\"4 1\",\"pages\":\"47\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12334418/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of rare diseases (Berlin, Germany)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s44162-025-00110-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of rare diseases (Berlin, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s44162-025-00110-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/8 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Severe clinical manifestation of mitochondrial disease due to the m.3243A>T variant: a case report of early-onset, multi-organ involvement and premature death.
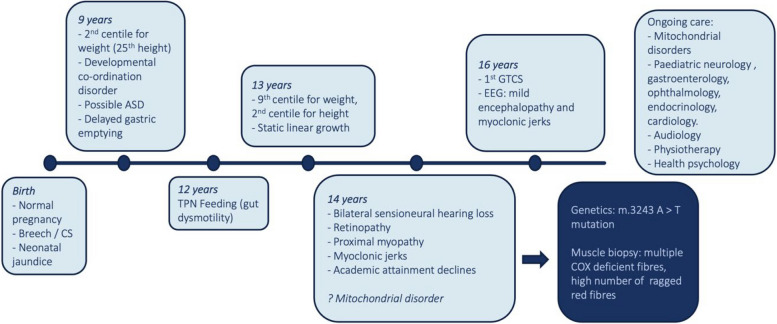
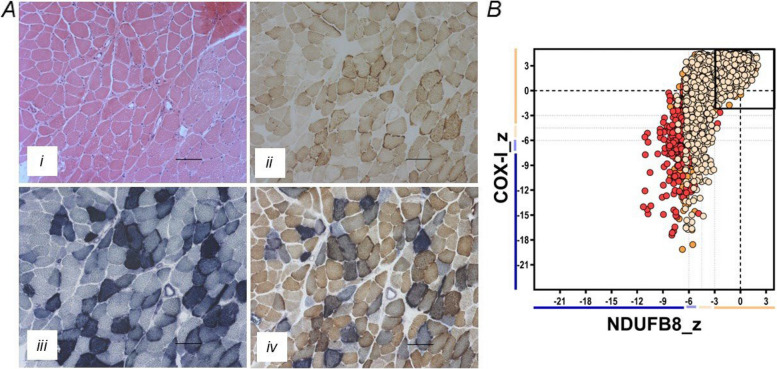
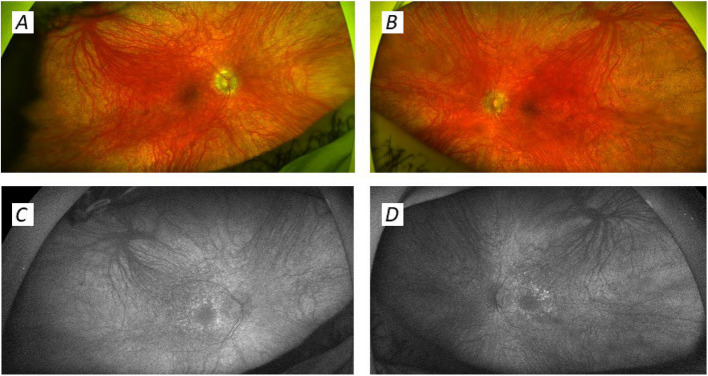
The spectrum of disease associated with pathogenic mitochondrial DNA (mtDNA) variants is wide. Most often, heteroplasmic mitochondrial DNA disease is the result of an adenine to guanine transition at position 3243 of mtDNA (m.3243A > G) in the MT-TL1 gene encoding tRNALeu(UUR). Here, we present a case of a patient with a rarer m.3243A > T variant whose phenotype was severe and included delayed growth, developmental delay, myoclonic jerks and tonic-clonic seizures, progressive myopathy, cerebellar ataxia, severe malnutrition due to intestinal dysmotility despite naso-jejunal feeding requiring total parenteral nutrition, bilateral sensorineural hearing loss, and visual impairment, including bilateral cataracts requiring treatment and pigmentary retinopathy. At age 18 years, he developed severe nephrotic syndrome secondary to a membranoproliferative pattern of glomerular injury, which was resistant to treatment and led to premature death.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


