{"title":"远端关节挛缩伴本体感觉和触觉受损(DAIPT)隐性形式中PIEZO2新内含子变异的功能表征。","authors":"Michela Bellardita, Ferruccio Romano, Ludovica Menta, Joana Soraia Martinheira Da Silva, Marzia Ognibene, Simona Baldassari, Marco Di Duca, Chiara Panicucci, Serena Baratto, Noemi Brolatti, Marina Pedemonte, Chiara Fiorillo, Claudio Bruno, Marcello Scala, Federico Zara, Francesca Faravelli, Francesca Madia, Serena Cappato, Renata Bocciardi, Valeria Capra","doi":"10.1002/mgg3.70126","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Distal arthrogryposis with impaired proprioception and touch (DAIPT) is a rare autosomal recessive neurological disease characterized by progressive alteration of mechanosensation. DAIPT is caused by loss of function variants in the PIEZO2 gene that encodes an ionic channel involved in mechanotransduction signaling. Our study started from the case of an 11-year-old boy with skeletal and neuromuscular features suggestive of DAIPT.</p><p><strong>Methods: </strong>Exome sequencing was performed on the trio. The identified variants in PIEZO2 were validated by Sanger sequencing. Functional assays of the variants were performed by minigene assay in HEK-293 cells and on patient-derived cells using NMD inhibitors.</p><p><strong>Results: </strong>Trio exome sequencing revealed the presence of two novel variants in the PIEZO2 gene: a nonsense variant (c.1924G>T; p.Glu642*) and an intronic variant of uncertain significance (c.2170-15A>G). Functional analysis demonstrated that the intronic variant disrupts splicing, leading to premature stop codon formation and possible mRNA targeting to nonsense-mediated mRNA decay (NMD). Molecular study in patient-derived fibroblasts with specific NMD inhibitors shows that transcripts derived from both alleles are degraded by NMD, thus confirming the effect of the nonsense variant and enabling reclassification of the VUS.</p><p><strong>Conclusion: </strong>We present the phenotypic and genetic description of a patient with features suggestive of DAIPT carrying novel biallelic variants in PIEZO2, one of which could be reclassified as pathogenic after functional assays. This study also provides a detailed review of all the published patients with DAIPT and expands the phenotypic and genetic understanding of DAIPT, aiding in diagnosis, genetic counseling, and clinical management.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 8","pages":"e70126"},"PeriodicalIF":1.6000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12329762/pdf/","citationCount":"0","resultStr":"{\"title\":\"Functional Characterization of a Novel Intronic Variant in PIEZO2 in a Recessive Form of Distal Arthrogryposis With Impaired Proprioception and Touch (DAIPT).\",\"authors\":\"Michela Bellardita, Ferruccio Romano, Ludovica Menta, Joana Soraia Martinheira Da Silva, Marzia Ognibene, Simona Baldassari, Marco Di Duca, Chiara Panicucci, Serena Baratto, Noemi Brolatti, Marina Pedemonte, Chiara Fiorillo, Claudio Bruno, Marcello Scala, Federico Zara, Francesca Faravelli, Francesca Madia, Serena Cappato, Renata Bocciardi, Valeria Capra\",\"doi\":\"10.1002/mgg3.70126\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Distal arthrogryposis with impaired proprioception and touch (DAIPT) is a rare autosomal recessive neurological disease characterized by progressive alteration of mechanosensation. DAIPT is caused by loss of function variants in the PIEZO2 gene that encodes an ionic channel involved in mechanotransduction signaling. Our study started from the case of an 11-year-old boy with skeletal and neuromuscular features suggestive of DAIPT.</p><p><strong>Methods: </strong>Exome sequencing was performed on the trio. The identified variants in PIEZO2 were validated by Sanger sequencing. Functional assays of the variants were performed by minigene assay in HEK-293 cells and on patient-derived cells using NMD inhibitors.</p><p><strong>Results: </strong>Trio exome sequencing revealed the presence of two novel variants in the PIEZO2 gene: a nonsense variant (c.1924G>T; p.Glu642*) and an intronic variant of uncertain significance (c.2170-15A>G). Functional analysis demonstrated that the intronic variant disrupts splicing, leading to premature stop codon formation and possible mRNA targeting to nonsense-mediated mRNA decay (NMD). Molecular study in patient-derived fibroblasts with specific NMD inhibitors shows that transcripts derived from both alleles are degraded by NMD, thus confirming the effect of the nonsense variant and enabling reclassification of the VUS.</p><p><strong>Conclusion: </strong>We present the phenotypic and genetic description of a patient with features suggestive of DAIPT carrying novel biallelic variants in PIEZO2, one of which could be reclassified as pathogenic after functional assays. This study also provides a detailed review of all the published patients with DAIPT and expands the phenotypic and genetic understanding of DAIPT, aiding in diagnosis, genetic counseling, and clinical management.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 8\",\"pages\":\"e70126\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12329762/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70126\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70126","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Functional Characterization of a Novel Intronic Variant in PIEZO2 in a Recessive Form of Distal Arthrogryposis With Impaired Proprioception and Touch (DAIPT).
Background: Distal arthrogryposis with impaired proprioception and touch (DAIPT) is a rare autosomal recessive neurological disease characterized by progressive alteration of mechanosensation. DAIPT is caused by loss of function variants in the PIEZO2 gene that encodes an ionic channel involved in mechanotransduction signaling. Our study started from the case of an 11-year-old boy with skeletal and neuromuscular features suggestive of DAIPT.
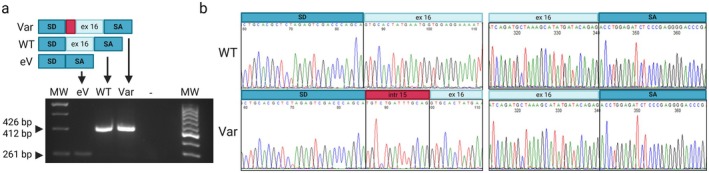
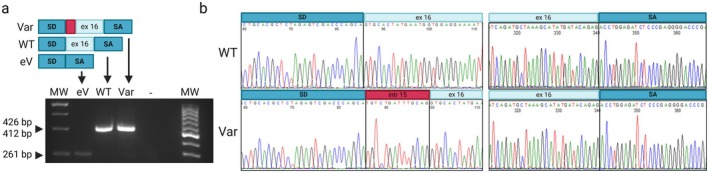
Methods: Exome sequencing was performed on the trio. The identified variants in PIEZO2 were validated by Sanger sequencing. Functional assays of the variants were performed by minigene assay in HEK-293 cells and on patient-derived cells using NMD inhibitors.
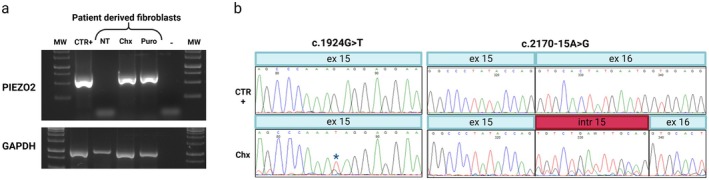
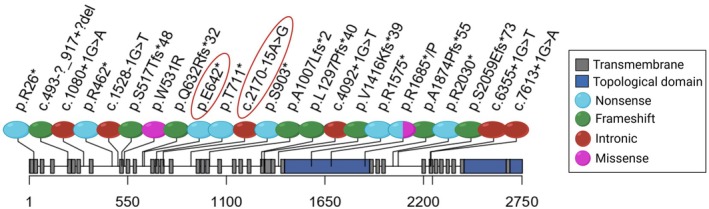
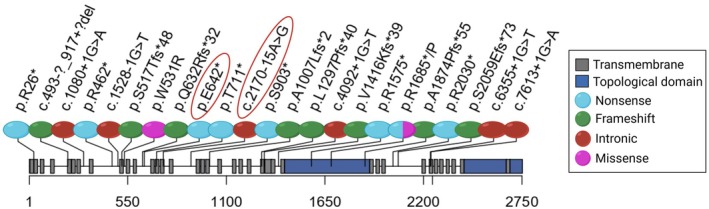
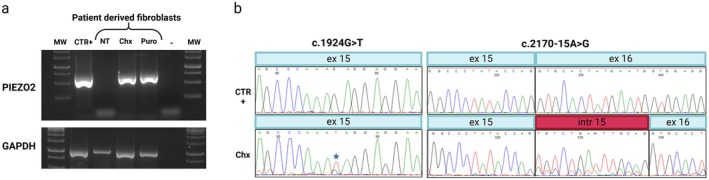
Results: Trio exome sequencing revealed the presence of two novel variants in the PIEZO2 gene: a nonsense variant (c.1924G>T; p.Glu642*) and an intronic variant of uncertain significance (c.2170-15A>G). Functional analysis demonstrated that the intronic variant disrupts splicing, leading to premature stop codon formation and possible mRNA targeting to nonsense-mediated mRNA decay (NMD). Molecular study in patient-derived fibroblasts with specific NMD inhibitors shows that transcripts derived from both alleles are degraded by NMD, thus confirming the effect of the nonsense variant and enabling reclassification of the VUS.
Conclusion: We present the phenotypic and genetic description of a patient with features suggestive of DAIPT carrying novel biallelic variants in PIEZO2, one of which could be reclassified as pathogenic after functional assays. This study also provides a detailed review of all the published patients with DAIPT and expands the phenotypic and genetic understanding of DAIPT, aiding in diagnosis, genetic counseling, and clinical management.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


