{"title":"非共价协同定向结晶(NSDC):一种多功能高能晶体的卤素工程策略","authors":"Cong Li, Zu-jia Lu, Chao Zhang, Mei-qi Xu, Bin-shan Zhao, Qi-yao Yu and Jian-guo Zhang","doi":"10.1039/D5CE00606F","DOIUrl":null,"url":null,"abstract":"<p >Achieving simultaneous optimization of energy density and safety performance remains a fundamental challenge in energetic materials science. Conventional design paradigms fail to address the cooperative regulation of weak intermolecular interactions governing crystal packing. Herein, we propose a halogen engineering strategy—noncovalent synergistic-directed crystallization (NSDC)—to decouple electronic effects from steric constraints for programmable control over solid-state architectures. Two heterocyclic systems, 1-(difluoromethyl)-5-methyl-4-nitro-1<em>H</em>-benzo[<em>d</em>][1,2,3]triazole (DFMNBT) and 3-iodo-4-nitropyrazole (INP), were designed to probe orthogonal noncovalent interaction networks. Single-crystal X-ray diffraction integrated with Hirshfeld surface analysis reveals that fluorine atoms direct antiparallel π–π stacking through dipole polarization (interlayer distance: 3.45–3.62 Å), while iodine atoms template lamellar growth <em>via</em> directional halogen bonding (I⋯O: 3.01 Å, <em>θ</em> = 178°). Density functional theory calculations indicate that, in the absence of strong hydrogen bonds between amino and nitro groups, fluorine substitution achieves high lattice energy through the synergistic effect of multiple weak interactions (111.08 <em>vs.</em> 125.71 kJ mol<small><sup>−1</sup></small> for TATB), whereas iodine incorporation constructs 3D frameworks through synergistic I⋯O halogen bonds (11.46 kJ mol<small><sup>−1</sup></small>) and halogen-π interactions, boosting lattice energy by 15.55% <em>versus</em> non-halogenated analogues. This work establishes a dual-pronged strategy for predictive engineering of multifunctional energetic crystals, resolving the long-standing energy-safety trade-off.</p>","PeriodicalId":70,"journal":{"name":"CrystEngComm","volume":" 31","pages":" 5346-5355"},"PeriodicalIF":2.6000,"publicationDate":"2025-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Noncovalent synergistic-directed crystallization (NSDC): a halogen engineering strategy for multifunctional energetic crystals†\",\"authors\":\"Cong Li, Zu-jia Lu, Chao Zhang, Mei-qi Xu, Bin-shan Zhao, Qi-yao Yu and Jian-guo Zhang\",\"doi\":\"10.1039/D5CE00606F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Achieving simultaneous optimization of energy density and safety performance remains a fundamental challenge in energetic materials science. Conventional design paradigms fail to address the cooperative regulation of weak intermolecular interactions governing crystal packing. Herein, we propose a halogen engineering strategy—noncovalent synergistic-directed crystallization (NSDC)—to decouple electronic effects from steric constraints for programmable control over solid-state architectures. Two heterocyclic systems, 1-(difluoromethyl)-5-methyl-4-nitro-1<em>H</em>-benzo[<em>d</em>][1,2,3]triazole (DFMNBT) and 3-iodo-4-nitropyrazole (INP), were designed to probe orthogonal noncovalent interaction networks. Single-crystal X-ray diffraction integrated with Hirshfeld surface analysis reveals that fluorine atoms direct antiparallel π–π stacking through dipole polarization (interlayer distance: 3.45–3.62 Å), while iodine atoms template lamellar growth <em>via</em> directional halogen bonding (I⋯O: 3.01 Å, <em>θ</em> = 178°). Density functional theory calculations indicate that, in the absence of strong hydrogen bonds between amino and nitro groups, fluorine substitution achieves high lattice energy through the synergistic effect of multiple weak interactions (111.08 <em>vs.</em> 125.71 kJ mol<small><sup>−1</sup></small> for TATB), whereas iodine incorporation constructs 3D frameworks through synergistic I⋯O halogen bonds (11.46 kJ mol<small><sup>−1</sup></small>) and halogen-π interactions, boosting lattice energy by 15.55% <em>versus</em> non-halogenated analogues. This work establishes a dual-pronged strategy for predictive engineering of multifunctional energetic crystals, resolving the long-standing energy-safety trade-off.</p>\",\"PeriodicalId\":70,\"journal\":{\"name\":\"CrystEngComm\",\"volume\":\" 31\",\"pages\":\" 5346-5355\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2025-07-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"CrystEngComm\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ce/d5ce00606f\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"CrystEngComm","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ce/d5ce00606f","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Noncovalent synergistic-directed crystallization (NSDC): a halogen engineering strategy for multifunctional energetic crystals†
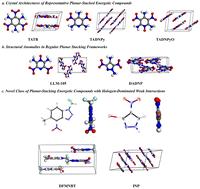
Achieving simultaneous optimization of energy density and safety performance remains a fundamental challenge in energetic materials science. Conventional design paradigms fail to address the cooperative regulation of weak intermolecular interactions governing crystal packing. Herein, we propose a halogen engineering strategy—noncovalent synergistic-directed crystallization (NSDC)—to decouple electronic effects from steric constraints for programmable control over solid-state architectures. Two heterocyclic systems, 1-(difluoromethyl)-5-methyl-4-nitro-1H-benzo[d][1,2,3]triazole (DFMNBT) and 3-iodo-4-nitropyrazole (INP), were designed to probe orthogonal noncovalent interaction networks. Single-crystal X-ray diffraction integrated with Hirshfeld surface analysis reveals that fluorine atoms direct antiparallel π–π stacking through dipole polarization (interlayer distance: 3.45–3.62 Å), while iodine atoms template lamellar growth via directional halogen bonding (I⋯O: 3.01 Å, θ = 178°). Density functional theory calculations indicate that, in the absence of strong hydrogen bonds between amino and nitro groups, fluorine substitution achieves high lattice energy through the synergistic effect of multiple weak interactions (111.08 vs. 125.71 kJ mol−1 for TATB), whereas iodine incorporation constructs 3D frameworks through synergistic I⋯O halogen bonds (11.46 kJ mol−1) and halogen-π interactions, boosting lattice energy by 15.55% versus non-halogenated analogues. This work establishes a dual-pronged strategy for predictive engineering of multifunctional energetic crystals, resolving the long-standing energy-safety trade-off.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


