单层MX2 (M=Mo, W;X=S, Se, Te),通过第一性原理计算
IF 2.4
4区 物理与天体物理
Q3 PHYSICS, CONDENSED MATTER
引用次数: 0
摘要
近年来,非中心对称材料中的二维(2D)体光伏效应(BPVE)由于其在高效二维太阳能电池和光电子领域的潜力而引起了人们的广泛关注。转移电流是BPVE的关键机制,对于设计增强的光伏性能至关重要。然而,这些材料的固有位移电流尚未得到全面而明确的研究。本文利用紧密结合模型分析和第一性原理计算,研究了缺乏自发极化的单层MX2(其中M = Mo, W, X = S, Se, Te)材料中的移位电流。我们构建了基于局部wannier90函数的紧密结合模型,包括3个(dz2, dx2−y2和dxy), 5个,7个,9个和11个带(没有或有自旋轨道耦合)来探索单层MX2的带结构。我们的研究结果表明,增加拟合频带的数量可以提高模型的准确性,尽管较少的频带仍然可以产生有意义的近似值。具体而言,S和Se化合物的位移电流在可见光范围内呈现明显的双峰结构,而Te化合物的位移电流在可见光范围内没有明显的峰结构。通过分析态的偏密度,我们发现双峰结构对应于Mo原子的dx2−y2和dxy轨道的峰值位置,以及S原子的py和px轨道的峰值位置。此外,双峰的位置与状态的节理密度不连续点重合。我们的研究阐明了态的能量密度,了解了位移电流峰值的来源,为二维体光伏效应的设计提供了理论依据。本文章由计算机程序翻译,如有差异,请以英文原文为准。
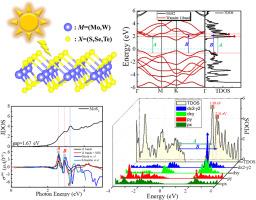
The shift current photovoltaic effect response in monolayer MX2 (M=Mo, W; X=S, Se, Te) via first-principles calculation
Recently, the two-dimensional (2D) bulk photovoltaic effect (BPVE) in non-centrosymmetric materials has garnered significant attention due to its potential for highly efficient 2D solar cells and optoelectronics. The shift current, a key mechanism underlying the BPVE, is crucial for designing the enhanced photovoltaic performance. However, the intrinsic shift current in these materials has not yet been comprehensively and unambiguously studied. Here, we investigate the shift current in monolayers MX2 (where M = Mo, W, and X = S, Se, Te) materials, which lack spontaneous polarization, using both tight-binding model analysis and first-principles calculations. We construct a tight-binding model based on the local wannier90 functions, incorporating three (dz2, dx2−y2, and dxy), five, seven, nine, and eleven bands (without or with spin-orbit coupling) to explore the band structure of monolayer MX2. Our findings indicate that increasing the fitted number of bands improves the accuracy of the model, although fewer bands can still yield meaningful approximations. Specifically, the shift current of the S and Se compounds exhibits a distinct double-peak structure within the visible light range, whereas the Te compounds display no obvious peak structure. By analyzing the partial density of states, we find that the double-peak structure corresponds to the peak positions of the Mo atom's dx2−y2 and dxy orbitals, as well as the S atom's py and px orbitals. Additionally, the location of the double peak coincides with the discontinuity point of the joint density of states. Our study elucidates the energy density of states to understand the origin of the shift current peak and offers a theoretical basis for the design of 2D bulk photovoltaic effects.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Solid State Communications
物理-物理:凝聚态物理
CiteScore
3.40
自引率
4.80%
发文量
287
审稿时长
51 days
期刊介绍:
Solid State Communications is an international medium for the publication of short communications and original research articles on significant developments in condensed matter science, giving scientists immediate access to important, recently completed work. The journal publishes original experimental and theoretical research on the physical and chemical properties of solids and other condensed systems and also on their preparation. The submission of manuscripts reporting research on the basic physics of materials science and devices, as well as of state-of-the-art microstructures and nanostructures, is encouraged.
A coherent quantitative treatment emphasizing new physics is expected rather than a simple accumulation of experimental data. Consistent with these aims, the short communications should be kept concise and short, usually not longer than six printed pages. The number of figures and tables should also be kept to a minimum. Solid State Communications now also welcomes original research articles without length restrictions.
The Fast-Track section of Solid State Communications is the venue for very rapid publication of short communications on significant developments in condensed matter science. The goal is to offer the broad condensed matter community quick and immediate access to publish recently completed papers in research areas that are rapidly evolving and in which there are developments with great potential impact.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


