新候选Sars-Cov-2 3CLpro(主要蛋白酶)抑制剂的开发:分子对接和分子动力学模拟研究
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
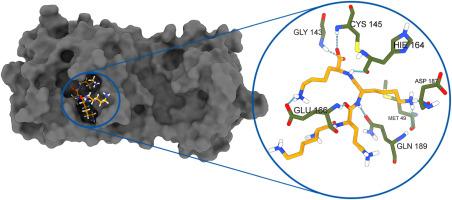
尽管COVID-19大流行导致全球人类流动正常化,但随着新变体的出现,该疾病仍然是一个主要威胁,因此它是药物开发的热门目标。3CLpro是SARS-CoV-2最关键的靶标之一,它是将病毒多蛋白切割成病毒复制所必需的非结构蛋白所必需的。通过分子对接,从ZINC15数据库中筛选出756958个分子与3CLpro进行对抗。前100位配体的对接分数在−82.25 kcal/mol和−98.06 kcal/mol之间。得分最高的9个配体和共结晶的N3进行了分子动力学模拟和结合自由能研究。配体与N3的结合自由能分别为- 22.04 kcal/mol和- 105.01 kcal/mol。此外,新研究的配体的结合自由能结果优于N3配体。此外,对接和MD结果表明配体与3CLpro结合位点关键残基如His41、Cys145和Glu166具有重要的相互作用。配体H03在对接自由能和结合自由能的计算上均优于所研究的配体N3。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Development of new inhibitor candidates for Sars-Cov-2 3CLpro (Main Protease): A molecular docking and molecular dynamics simulation study
Despite the normalization in the human mobility worldwide caused by COVID-19 pandemics, the disease still remains as a major threat with emerge of new variants and therefore it is a popular target for the drug development. The 3CLpro is one of the most pivotal targets of SARS-CoV-2 which is needed for the cleavage of viral polyproteins into nonstructural proteins that are essential for viral replication. A total of 756958 molecules from ZINC15 database have been screened against 3CLpro by molecular docking. The docking scores of the top 100 ligands were in the range of −82.25 kcal/mol and −98.06 kcal/mol. Top scored nine ligands and co-crystalized N3 were forwarded to the MD simulations and binding free energy studies. The binding free energy values of ligands and N3 were in the range of −22.04 kcal/mol and −105.01 kcal/mol. Moreover, the binding free energy results of newly studied ligands were better than N3 ligand. In addition, the docking and the MD results indicated that ligands have valuable interactions with 3CLpro binding site key residues, such as His41, Cys145 and Glu166. The ligand H03 demonstrated better results than all studied ligands as well than N3 for both of docking and binding free energy calculations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


