Joël Wellauer, Michael L. Pattuwage, Egan H. Doeven, Timothy U. Connell, Oliver S. Wenger* and Paul S. Francis*,
{"title":"再思考铁(III)配合物在LMCT光氧化还原催化中的激发态氧化还原特性。","authors":"Joël Wellauer, Michael L. Pattuwage, Egan H. Doeven, Timothy U. Connell, Oliver S. Wenger* and Paul S. Francis*, ","doi":"10.1021/jacs.5c08841","DOIUrl":null,"url":null,"abstract":"<p >The reduction potentials of electronically excited states are crucial input values for photoredox reaction design. Since they are not directly measurable, they are typically estimated from the corresponding ground-state potentials and excited-state energies. Here, we demonstrate that this commonly applied approach breaks down for low-spin d<sup>5</sup> complexes of iron(III) with photoactive ligand-to-metal charge transfer (LMCT) excited states. Stern–Volmer luminescence quenching, photocatalytic experiments, and detailed thermodynamic analyses demonstrate that the true potentials for the oxidation of excited-state iron(III) complexes are up to 0.7 V lower than anticipated, resulting in a nearly 70 kJ/mol change in the driving forces of photoinduced electron transfer reactions. Our analysis further indicates that other complexes with LMCT-excited states and partially filled d-orbitals are likely to exhibit the same behavior, because LMCT-excited-state quenching removes the highest-energy electron from the t<sub>2g</sub> orbital but results in a formally ligand-centered oxidation, whereas the first ground-state oxidation is typically metal-centered. These findings have significant implications for the use of the emerging class of complexes with photoactive LMCT-excited states as well as for the broader field of LMCT photoredox catalysis in synthetic chemistry.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"147 32","pages":"29304–29314"},"PeriodicalIF":15.6000,"publicationDate":"2025-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Rethinking the Excited-State Redox Properties of Iron(III) Complexes for LMCT Photoredox Catalysis\",\"authors\":\"Joël Wellauer, Michael L. Pattuwage, Egan H. Doeven, Timothy U. Connell, Oliver S. Wenger* and Paul S. Francis*, \",\"doi\":\"10.1021/jacs.5c08841\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The reduction potentials of electronically excited states are crucial input values for photoredox reaction design. Since they are not directly measurable, they are typically estimated from the corresponding ground-state potentials and excited-state energies. Here, we demonstrate that this commonly applied approach breaks down for low-spin d<sup>5</sup> complexes of iron(III) with photoactive ligand-to-metal charge transfer (LMCT) excited states. Stern–Volmer luminescence quenching, photocatalytic experiments, and detailed thermodynamic analyses demonstrate that the true potentials for the oxidation of excited-state iron(III) complexes are up to 0.7 V lower than anticipated, resulting in a nearly 70 kJ/mol change in the driving forces of photoinduced electron transfer reactions. Our analysis further indicates that other complexes with LMCT-excited states and partially filled d-orbitals are likely to exhibit the same behavior, because LMCT-excited-state quenching removes the highest-energy electron from the t<sub>2g</sub> orbital but results in a formally ligand-centered oxidation, whereas the first ground-state oxidation is typically metal-centered. These findings have significant implications for the use of the emerging class of complexes with photoactive LMCT-excited states as well as for the broader field of LMCT photoredox catalysis in synthetic chemistry.</p>\",\"PeriodicalId\":49,\"journal\":{\"name\":\"Journal of the American Chemical Society\",\"volume\":\"147 32\",\"pages\":\"29304–29314\"},\"PeriodicalIF\":15.6000,\"publicationDate\":\"2025-08-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jacs.5c08841\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.5c08841","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Rethinking the Excited-State Redox Properties of Iron(III) Complexes for LMCT Photoredox Catalysis
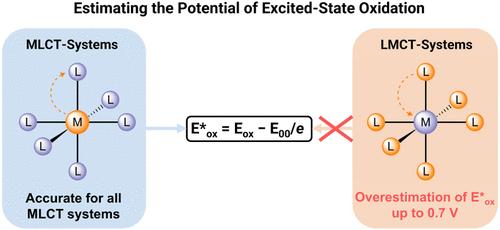
The reduction potentials of electronically excited states are crucial input values for photoredox reaction design. Since they are not directly measurable, they are typically estimated from the corresponding ground-state potentials and excited-state energies. Here, we demonstrate that this commonly applied approach breaks down for low-spin d5 complexes of iron(III) with photoactive ligand-to-metal charge transfer (LMCT) excited states. Stern–Volmer luminescence quenching, photocatalytic experiments, and detailed thermodynamic analyses demonstrate that the true potentials for the oxidation of excited-state iron(III) complexes are up to 0.7 V lower than anticipated, resulting in a nearly 70 kJ/mol change in the driving forces of photoinduced electron transfer reactions. Our analysis further indicates that other complexes with LMCT-excited states and partially filled d-orbitals are likely to exhibit the same behavior, because LMCT-excited-state quenching removes the highest-energy electron from the t2g orbital but results in a formally ligand-centered oxidation, whereas the first ground-state oxidation is typically metal-centered. These findings have significant implications for the use of the emerging class of complexes with photoactive LMCT-excited states as well as for the broader field of LMCT photoredox catalysis in synthetic chemistry.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


