{"title":"生物膜中的离子-脂质相互作用:来自分子动力学和量子化学计算的见解。","authors":"Subhasish Mallick*, ","doi":"10.1021/acs.jpcb.5c03120","DOIUrl":null,"url":null,"abstract":"<p >Understanding ion–lipid interactions at biomembrane interfaces is fundamental to deciphering biological processes and designing biomimetic systems. While classical MD simulations provide valuable insights into ion–lipid interactions, they sometimes fail to reproduce experimental observations due to the limitations inherent in the force field accuracy. In this study, we employ high-level ab initio calculations along with molecular dynamics (MD) simulations to elucidate the interplay between cations (Na<sup>+</sup>, K<sup>+</sup>, Ca<sup>2+</sup>, and Mg<sup>2+</sup>), as well as Cl<sup>–</sup> counterions, with phosphatidylcholine lipid head groups. Optimized configurations reveal that Na<sup>+</sup> and K<sup>+</sup> preferentially interact with phosphatic oxygen (O<sub>P</sub>) rather than carbonyl oxygen (O<sub>C</sub>), whereas Ca<sup>2+</sup> and Mg<sup>2+</sup> exhibit dual-site binding at O<sub>P</sub> and O<sub>C</sub>. Notably, all cations except Mg<sup>2+</sup> bind directly to the lipid head groups by shedding hydration water. Due to its high hydration energy, Mg<sup>2+</sup> retains its hexahydrated state, interacting indirectly with lipid head groups via a bridging water molecule. Similarly, Cl<sup>–</sup> avoids direct interactions with choline groups and instead binds via water-mediated bridges. Energetic analysis at the DLPNO–CCSD(T)/cc-pVTZ//B3LYP-D3/6–31G** level of theory reveals that Na<sup>+</sup> exhibits interaction energies significantly weaker than those of Ca<sup>2+</sup>, aligning well with experimental observations, whereas classical MD often overestimates the binding affinity of Na<sup>+</sup>. A trend of increasing binding strength with lipid cluster size is observed, with divalent cations displaying a biphasic binding trend, where the largest increase in interaction energy occurs between monomers and dimers, suggesting near-saturation of binding sites in dimeric lipid clusters. This observation aligns with experimental findings that Ca<sup>2+</sup> preferentially bridges two lipid headgroups, a feature often misrepresented in classical force fields. These findings provide crucial insights into ion–lipid interactions in biological membranes, highlighting the limitations of classical force fields and emphasizing the need to incorporate ab initio-derived parameters to improve the accuracy of MD simulations.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"129 32","pages":"8156–8165"},"PeriodicalIF":2.9000,"publicationDate":"2025-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ion–Lipid Interactions in Biological Membranes: Insights from Combined Molecular Dynamics and Quantum Chemical Calculations\",\"authors\":\"Subhasish Mallick*, \",\"doi\":\"10.1021/acs.jpcb.5c03120\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Understanding ion–lipid interactions at biomembrane interfaces is fundamental to deciphering biological processes and designing biomimetic systems. While classical MD simulations provide valuable insights into ion–lipid interactions, they sometimes fail to reproduce experimental observations due to the limitations inherent in the force field accuracy. In this study, we employ high-level ab initio calculations along with molecular dynamics (MD) simulations to elucidate the interplay between cations (Na<sup>+</sup>, K<sup>+</sup>, Ca<sup>2+</sup>, and Mg<sup>2+</sup>), as well as Cl<sup>–</sup> counterions, with phosphatidylcholine lipid head groups. Optimized configurations reveal that Na<sup>+</sup> and K<sup>+</sup> preferentially interact with phosphatic oxygen (O<sub>P</sub>) rather than carbonyl oxygen (O<sub>C</sub>), whereas Ca<sup>2+</sup> and Mg<sup>2+</sup> exhibit dual-site binding at O<sub>P</sub> and O<sub>C</sub>. Notably, all cations except Mg<sup>2+</sup> bind directly to the lipid head groups by shedding hydration water. Due to its high hydration energy, Mg<sup>2+</sup> retains its hexahydrated state, interacting indirectly with lipid head groups via a bridging water molecule. Similarly, Cl<sup>–</sup> avoids direct interactions with choline groups and instead binds via water-mediated bridges. Energetic analysis at the DLPNO–CCSD(T)/cc-pVTZ//B3LYP-D3/6–31G** level of theory reveals that Na<sup>+</sup> exhibits interaction energies significantly weaker than those of Ca<sup>2+</sup>, aligning well with experimental observations, whereas classical MD often overestimates the binding affinity of Na<sup>+</sup>. A trend of increasing binding strength with lipid cluster size is observed, with divalent cations displaying a biphasic binding trend, where the largest increase in interaction energy occurs between monomers and dimers, suggesting near-saturation of binding sites in dimeric lipid clusters. This observation aligns with experimental findings that Ca<sup>2+</sup> preferentially bridges two lipid headgroups, a feature often misrepresented in classical force fields. These findings provide crucial insights into ion–lipid interactions in biological membranes, highlighting the limitations of classical force fields and emphasizing the need to incorporate ab initio-derived parameters to improve the accuracy of MD simulations.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\"129 32\",\"pages\":\"8156–8165\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-07-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c03120\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c03120","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Ion–Lipid Interactions in Biological Membranes: Insights from Combined Molecular Dynamics and Quantum Chemical Calculations
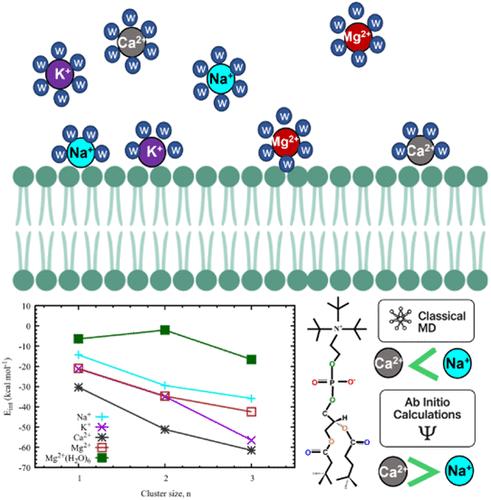
Understanding ion–lipid interactions at biomembrane interfaces is fundamental to deciphering biological processes and designing biomimetic systems. While classical MD simulations provide valuable insights into ion–lipid interactions, they sometimes fail to reproduce experimental observations due to the limitations inherent in the force field accuracy. In this study, we employ high-level ab initio calculations along with molecular dynamics (MD) simulations to elucidate the interplay between cations (Na+, K+, Ca2+, and Mg2+), as well as Cl– counterions, with phosphatidylcholine lipid head groups. Optimized configurations reveal that Na+ and K+ preferentially interact with phosphatic oxygen (OP) rather than carbonyl oxygen (OC), whereas Ca2+ and Mg2+ exhibit dual-site binding at OP and OC. Notably, all cations except Mg2+ bind directly to the lipid head groups by shedding hydration water. Due to its high hydration energy, Mg2+ retains its hexahydrated state, interacting indirectly with lipid head groups via a bridging water molecule. Similarly, Cl– avoids direct interactions with choline groups and instead binds via water-mediated bridges. Energetic analysis at the DLPNO–CCSD(T)/cc-pVTZ//B3LYP-D3/6–31G** level of theory reveals that Na+ exhibits interaction energies significantly weaker than those of Ca2+, aligning well with experimental observations, whereas classical MD often overestimates the binding affinity of Na+. A trend of increasing binding strength with lipid cluster size is observed, with divalent cations displaying a biphasic binding trend, where the largest increase in interaction energy occurs between monomers and dimers, suggesting near-saturation of binding sites in dimeric lipid clusters. This observation aligns with experimental findings that Ca2+ preferentially bridges two lipid headgroups, a feature often misrepresented in classical force fields. These findings provide crucial insights into ion–lipid interactions in biological membranes, highlighting the limitations of classical force fields and emphasizing the need to incorporate ab initio-derived parameters to improve the accuracy of MD simulations.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


