丙二酸二甲酯通过抑制fth1介导的铁蛋白吞噬来保护新生儿缺氧缺血后的大脑和神经行为表型
IF 11.9
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
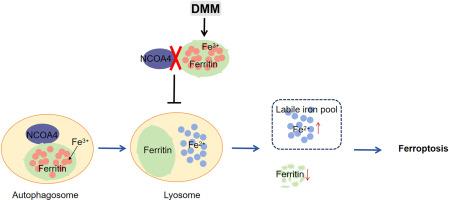
背景:缺氧缺血性脑损伤(HIBD)是新生儿神经元损伤和死亡的主要原因。目前的预防和治疗干预措施显示有限的临床疗效。新出现的证据表明,铁下垂是HIBD病理生理中的一个关键机制,将其定位为一个有希望的治疗靶点。丙二酸二甲酯(DMM)是琥珀酸脱氢酶的竞争性抑制剂,在多种神经系统疾病模型中显示出神经保护作用。然而,DMM对新生儿HIBD的影响尚未得到研究。目的探讨DMM对新生儿HIBD的神经保护作用,并阐明其作用机制。方法建立新生雄性C57BL/6J小鼠HIBD模型,给予不同剂量的DMM或对照。定量评估包括脑梗死体积测量、神经元尼氏染色、神经行为、亚铁离子(Fe2+)、丙二醛(MDA)水平、4-羟基壬烯醛(4- hne)表达、溶质载体家族7成员11 (SLC7A11,系统Xc−)/谷胱甘肽过氧化物酶4 (GPX4)抗氧化轴表达水平。体外平行研究采用氧-葡萄糖剥夺/再灌注处理HT22细胞,探讨DMM对铁凋亡的影响及其潜在机制。western blotting分析了核受体共激活因子4 (NCOA4)、SQSTM1/p62、铁蛋白重链1 (FTH1)、微管相关蛋白轻3II (LC3II)等铁蛋白自噬的关键因子。采用共免疫沉淀法(Co-IP)分析了dmm处理的HIBD小鼠脑皮质组织中NCOA4与FTH1的分子相互作用。在细胞模型中通过Fth1敲低进一步研究DMM对铁下垂的调节。免疫荧光染色评价DMM在细胞器水平抑制铁蛋白降解和溶酶体铁2+积累的能力。结果dmm治疗在HIBD模型中显示出神经保护作用,与对照组相比,新生小鼠脑梗死体积减少,nsli阳性神经元数量增加,认知和运动功能改善。此外,DMM干预显著调节脑皮质组织和HT22细胞中与铁中毒相关的生物标志物,减少亚铁离子(Fe2+)积累,减少脂质过氧化产物(MDA和4-HNE),增强SLC7A11/GPX4抗氧化系统活性。重要的是,DMM特异性调节核心铁蛋白自噬成分:抑制NCOA4和LC3II的表达,同时上调FTH1和p62的水平。Co-IP揭示了DMM在机制上破坏了NCOA4和FTH1之间的蛋白相互作用,有效地抑制了铁蛋白自噬的进展。抗铁沉的作用依赖于Fth1,正如在体外敲除Fth1后DMM保护作用的逆转所证明的那样。免疫荧光分析显示,DMM降低了fth1 -溶酶体相关膜蛋白2的共定位,FerroOrange/LysoTracker Green双染色证实其抑制溶酶体铁积累,共同表明DMM在细胞器水平上调控。结论dmm通过特异性靶向FTH1,破坏NCOA4-FTH1相互作用,抑制铁中毒诱导的神经元死亡,从而减轻HIBD。这些发现使DMM成为新生儿缺氧缺血性脑病临床治疗的有希望的治疗候选药物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Dimethyl malonate preserves brain and neurobehavioral phenotype following neonatal hypoxia–ischemia by inhibiting FTH1-mediated ferritinophagy
Background
Hypoxic–ischemic brain damage (HIBD) is a predominant cause of neuronal injury and mortality in newborns. Current preventive and therapeutic interventions demonstrate limited clinical efficacy. Emerging evidence reveals ferroptosis as a critical mechanism within HIBD pathophysiology, positioning it as a promising therapeutic target. Dimethyl malonate (DMM), a competitive inhibitor of succinate dehydrogenase, has demonstrated neuroprotective properties across multiple models of neurological disorders. However, the impact of DMM on the neonatal HIBD has not been studied.
Aim
To investigate the neuroprotective effects of DMM against neonatal HIBD and elucidate its mechanisms of action.
Methods
We created a model of HIBD in neonatal male C57BL/6J mice and administered various doses of DMM or vehicle control. Quantitative assessments included cerebral infarct volume measurement, Nissl staining for neurons, neurological behavior, ferrous ion (Fe2+), malondialdehyde (MDA) level, 4-hydroxynonenal (4-HNE) expression, and solute carrier family 7 member 11 (SLC7A11, system Xc−)/glutathione peroxidase 4 (GPX4) antioxidant axis expression level. Parallel studies in vitro employed oxygen-glucose deprivation/reperfusion-treated HT22 cells to investigate the effects of DMM on ferroptosis and its underlying mechanisms. Moreover, key factors of ferritinophagy, including nuclear receptor coactivator 4 (NCOA4), SQSTM1/p62, ferritin heavy chain 1 (FTH1), and microtubule-associated protein light 3 II (LC3II) were analyzed by western blotting. Molecular interactions between NCOA4 and FTH1 in brain cortical tissues of DMM-treated HIBD mice were analyzed by coimmunoprecipitation (Co-IP). Ferroptosis regulation by DMM was further investigated via Fth1 knockdown in cellular models. Immunofluorescence staining was used to evaluate the capacity of DMM to suppress ferritin degradation and lysosomal Fe2+ accumulation at the organelle level.
Results
DMM treatment demonstrated its neuroprotective efficacy in HIBD models, as evidenced by a reduction in cerebral infarct volume, an increase in the number of Nissl-positive neurons, and improved cognitive and motor functions in neonatal mice compared with controls. Additionally, the DMM intervention significantly modulated ferroptosis-related biomarkers in brain cortical tissues and HT22 cells, decreasing ferrous ion (Fe2+) accumulation, reducing lipid peroxidation products (MDA and 4-HNE), and enhancing SLC7A11/GPX4 antioxidant system activity. Importantly, DMM specifically regulated core ferritinophagy components: suppressing NCOA4 and LC3II expression while upregulating FTH1 and p62 levels. Co-IP revealed that mechanistically, DMM disrupted the protein interaction between NCOA4 and FTH1, effectively inhibiting ferritinophagy progression. The effects of antiferroptosis were FTH1-dependent, as demonstrated by reversal of the DMM protective effect following Fth1 knockdown in vitro. Immunofluorescence analysis showed that DMM decreased the colocalization of FTH1-lysosome-associated membrane protein 2, and FerroOrange/LysoTracker Green dual staining confirmed its inhibition of lysosomal iron accumulation, collectively indicating regulation of DMM at the organelle level.
Conclusion
DMM suppressed ferroptosis-induced neuronal death by specifically targeting FTH1 and disrupting the NCOA4-FTH1 interaction, thereby mitigating HIBD. These findings position DMM as a promising therapeutic candidate for the clinical management of neonatal hypoxic–ischemic encephalopathy.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Redox Biology
BIOCHEMISTRY & MOLECULAR BIOLOGY-
CiteScore
19.90
自引率
3.50%
发文量
318
审稿时长
25 days
期刊介绍:
Redox Biology is the official journal of the Society for Redox Biology and Medicine and the Society for Free Radical Research-Europe. It is also affiliated with the International Society for Free Radical Research (SFRRI). This journal serves as a platform for publishing pioneering research, innovative methods, and comprehensive review articles in the field of redox biology, encompassing both health and disease.
Redox Biology welcomes various forms of contributions, including research articles (short or full communications), methods, mini-reviews, and commentaries. Through its diverse range of published content, Redox Biology aims to foster advancements and insights in the understanding of redox biology and its implications.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


