{"title":"2,3-二氟吡啶的氟位置依赖电离动力学和结构畸变:vv - mati和理论研究","authors":"Hyojung Kim, Sung Man Park and Chan Ho Kwon","doi":"10.1039/D5CP02425K","DOIUrl":null,"url":null,"abstract":"<p >Pyridine derivatives are integral to fields such as organic chemistry, materials science, and pharmaceuticals owing to their distinct electronic and structural properties. While fluorine substitution on the pyridine ring is known to modulate these properties, the specific effects of fluorine positioning—particularly at the <em>ortho</em> and <em>meta</em> positions—on the molecular orbitals, ionization energies, and cationic structures remain insufficiently understood. In this study, we investigate the ionization-induced structural dynamics of 2,3-difluoropyridine (2,3-DFP), which incorporates both <em>ortho</em>- and <em>meta</em>-fluorine substituents, using high-resolution vacuum ultraviolet mass-analyzed threshold ionization (VUV-MATI) spectroscopy in conjunction with Franck–Condon (FC) simulations and natural bond orbital analysis. Precise measurement of the adiabatic ionization energy (AIE) yields a value of 9.6958 ± 0.0004 eV for 2,3-DFP, which is lower than that of 2,6-DFP due to weaker hyperconjugative stabilization of the highest occupied molecular orbital (HOMO) by <em>meta</em>-fluorine. The VUV-MATI spectrum reveals significant geometric distortion upon ionization, notably in out-of-plane vibrational modes, which become FC-active due to symmetry lowering. FC fitting with a refined, slightly nonplanar geometry closely reproduces the experimental spectrum. Additional unassigned peaks are attributed to the D<small><sub>1</sub></small> state, originating from ionization of the HOMO−1 nonbonding orbital, and a second AIE of 9.7643 ± 0.0004 eV is proposed. These findings clarify how fluorine substitution patterns modulate valence orbital energies, cationic structures, and vibrational dynamics. This work provides a detailed framework for understanding the stereoelectronic effects of fluorination in heteroaromatic systems and offers practical insight for designing functional materials and pharmaceuticals with tunable electronic properties.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 33","pages":" 17399-17406"},"PeriodicalIF":2.9000,"publicationDate":"2025-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fluorine position-dependent ionization dynamics and structural distortion in 2,3-difluoropyridine: a VUV-MATI and theoretical study\",\"authors\":\"Hyojung Kim, Sung Man Park and Chan Ho Kwon\",\"doi\":\"10.1039/D5CP02425K\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Pyridine derivatives are integral to fields such as organic chemistry, materials science, and pharmaceuticals owing to their distinct electronic and structural properties. While fluorine substitution on the pyridine ring is known to modulate these properties, the specific effects of fluorine positioning—particularly at the <em>ortho</em> and <em>meta</em> positions—on the molecular orbitals, ionization energies, and cationic structures remain insufficiently understood. In this study, we investigate the ionization-induced structural dynamics of 2,3-difluoropyridine (2,3-DFP), which incorporates both <em>ortho</em>- and <em>meta</em>-fluorine substituents, using high-resolution vacuum ultraviolet mass-analyzed threshold ionization (VUV-MATI) spectroscopy in conjunction with Franck–Condon (FC) simulations and natural bond orbital analysis. Precise measurement of the adiabatic ionization energy (AIE) yields a value of 9.6958 ± 0.0004 eV for 2,3-DFP, which is lower than that of 2,6-DFP due to weaker hyperconjugative stabilization of the highest occupied molecular orbital (HOMO) by <em>meta</em>-fluorine. The VUV-MATI spectrum reveals significant geometric distortion upon ionization, notably in out-of-plane vibrational modes, which become FC-active due to symmetry lowering. FC fitting with a refined, slightly nonplanar geometry closely reproduces the experimental spectrum. Additional unassigned peaks are attributed to the D<small><sub>1</sub></small> state, originating from ionization of the HOMO−1 nonbonding orbital, and a second AIE of 9.7643 ± 0.0004 eV is proposed. These findings clarify how fluorine substitution patterns modulate valence orbital energies, cationic structures, and vibrational dynamics. This work provides a detailed framework for understanding the stereoelectronic effects of fluorination in heteroaromatic systems and offers practical insight for designing functional materials and pharmaceuticals with tunable electronic properties.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 33\",\"pages\":\" 17399-17406\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-07-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02425k\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02425k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Fluorine position-dependent ionization dynamics and structural distortion in 2,3-difluoropyridine: a VUV-MATI and theoretical study
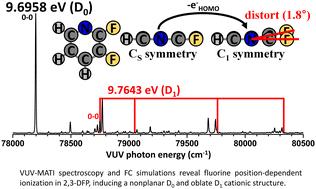
Pyridine derivatives are integral to fields such as organic chemistry, materials science, and pharmaceuticals owing to their distinct electronic and structural properties. While fluorine substitution on the pyridine ring is known to modulate these properties, the specific effects of fluorine positioning—particularly at the ortho and meta positions—on the molecular orbitals, ionization energies, and cationic structures remain insufficiently understood. In this study, we investigate the ionization-induced structural dynamics of 2,3-difluoropyridine (2,3-DFP), which incorporates both ortho- and meta-fluorine substituents, using high-resolution vacuum ultraviolet mass-analyzed threshold ionization (VUV-MATI) spectroscopy in conjunction with Franck–Condon (FC) simulations and natural bond orbital analysis. Precise measurement of the adiabatic ionization energy (AIE) yields a value of 9.6958 ± 0.0004 eV for 2,3-DFP, which is lower than that of 2,6-DFP due to weaker hyperconjugative stabilization of the highest occupied molecular orbital (HOMO) by meta-fluorine. The VUV-MATI spectrum reveals significant geometric distortion upon ionization, notably in out-of-plane vibrational modes, which become FC-active due to symmetry lowering. FC fitting with a refined, slightly nonplanar geometry closely reproduces the experimental spectrum. Additional unassigned peaks are attributed to the D1 state, originating from ionization of the HOMO−1 nonbonding orbital, and a second AIE of 9.7643 ± 0.0004 eV is proposed. These findings clarify how fluorine substitution patterns modulate valence orbital energies, cationic structures, and vibrational dynamics. This work provides a detailed framework for understanding the stereoelectronic effects of fluorination in heteroaromatic systems and offers practical insight for designing functional materials and pharmaceuticals with tunable electronic properties.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


