{"title":"单细胞RNA测序数据的软图聚类。","authors":"Ping Xu, Pengfei Wang, Zhiyuan Ning, Meng Xiao, Min Wu, Yuanchun Zhou","doi":"10.1186/s12859-025-06231-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Clustering analysis is fundamental in single-cell RNA sequencing (scRNA-seq) data analysis for elucidating cellular heterogeneity and diversity. Recent graph-based scRNA-seq clustering methods, particularly graph neural networks (GNNs), have significantly improved in tackling the challenges of high-dimension, high-sparsity, and frequent dropout events that lead to ambiguous cell population boundaries. However, one major challenge for GNN-based methods is their reliance on hard graph constructions derived from similarity matrices. These constructions introduce difficulties when applied to scRNA-seq data due to: (i) The simplification of intercellular relationships into binary edges (0 or 1) by applying thresholds, which restricts the capture of continuous similarity features among cells and leads to significant information loss. (ii) The presence of significant inter-cluster connections within hard graphs, which can confuse GNN methods that rely heavily on graph structures, potentially causing erroneous message propagation and biased clustering outcomes.</p><p><strong>Results: </strong>To tackle these challenges, we introduce scSGC, a Soft Graph Clustering for single-cell RNA sequencing data, which aims to more accurately characterize continuous similarities among cells through non-binary edge weights, thereby mitigating the limitations of rigid data structures. The scSGC framework comprises three core components: (i) a zero-inflated negative binomial (ZINB)-based feature autoencoder designed to effectively handle the sparsity and dropout issues in scRNA-seq data; (ii) a dual-channel cut-informed soft graph embedding module, constructed through deep graph-cut information, capturing continuous similarities between cells while preserving the intrinsic data structures of scRNA-seq; and (iii) an optimal transport-based clustering optimization module, achieving optimal delineation of cell populations while maintaining high biological relevance.</p><p><strong>Conclusion: </strong>By integrating dual-channel cut-informed soft graph representation learning, a ZINB-based feature autoencoder, and optimal transport-driven clustering optimization, scSGC effectively overcomes the challenges associated with traditional hard graph constructions in GNN methods. Extensive experiments across ten datasets demonstrate that scSGC outperforms 13 state-of-the-art clustering models in clustering accuracy, cell type annotation, and computational efficiency. These results highlight its substantial potential to advance scRNA-seq data analysis and deepen our understanding of cellular heterogeneity.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"195"},"PeriodicalIF":3.3000,"publicationDate":"2025-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12291377/pdf/","citationCount":"0","resultStr":"{\"title\":\"Soft graph clustering for single-cell RNA sequencing data.\",\"authors\":\"Ping Xu, Pengfei Wang, Zhiyuan Ning, Meng Xiao, Min Wu, Yuanchun Zhou\",\"doi\":\"10.1186/s12859-025-06231-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Clustering analysis is fundamental in single-cell RNA sequencing (scRNA-seq) data analysis for elucidating cellular heterogeneity and diversity. Recent graph-based scRNA-seq clustering methods, particularly graph neural networks (GNNs), have significantly improved in tackling the challenges of high-dimension, high-sparsity, and frequent dropout events that lead to ambiguous cell population boundaries. However, one major challenge for GNN-based methods is their reliance on hard graph constructions derived from similarity matrices. These constructions introduce difficulties when applied to scRNA-seq data due to: (i) The simplification of intercellular relationships into binary edges (0 or 1) by applying thresholds, which restricts the capture of continuous similarity features among cells and leads to significant information loss. (ii) The presence of significant inter-cluster connections within hard graphs, which can confuse GNN methods that rely heavily on graph structures, potentially causing erroneous message propagation and biased clustering outcomes.</p><p><strong>Results: </strong>To tackle these challenges, we introduce scSGC, a Soft Graph Clustering for single-cell RNA sequencing data, which aims to more accurately characterize continuous similarities among cells through non-binary edge weights, thereby mitigating the limitations of rigid data structures. The scSGC framework comprises three core components: (i) a zero-inflated negative binomial (ZINB)-based feature autoencoder designed to effectively handle the sparsity and dropout issues in scRNA-seq data; (ii) a dual-channel cut-informed soft graph embedding module, constructed through deep graph-cut information, capturing continuous similarities between cells while preserving the intrinsic data structures of scRNA-seq; and (iii) an optimal transport-based clustering optimization module, achieving optimal delineation of cell populations while maintaining high biological relevance.</p><p><strong>Conclusion: </strong>By integrating dual-channel cut-informed soft graph representation learning, a ZINB-based feature autoencoder, and optimal transport-driven clustering optimization, scSGC effectively overcomes the challenges associated with traditional hard graph constructions in GNN methods. Extensive experiments across ten datasets demonstrate that scSGC outperforms 13 state-of-the-art clustering models in clustering accuracy, cell type annotation, and computational efficiency. These results highlight its substantial potential to advance scRNA-seq data analysis and deepen our understanding of cellular heterogeneity.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"195\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12291377/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06231-z\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06231-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Soft graph clustering for single-cell RNA sequencing data.
Background: Clustering analysis is fundamental in single-cell RNA sequencing (scRNA-seq) data analysis for elucidating cellular heterogeneity and diversity. Recent graph-based scRNA-seq clustering methods, particularly graph neural networks (GNNs), have significantly improved in tackling the challenges of high-dimension, high-sparsity, and frequent dropout events that lead to ambiguous cell population boundaries. However, one major challenge for GNN-based methods is their reliance on hard graph constructions derived from similarity matrices. These constructions introduce difficulties when applied to scRNA-seq data due to: (i) The simplification of intercellular relationships into binary edges (0 or 1) by applying thresholds, which restricts the capture of continuous similarity features among cells and leads to significant information loss. (ii) The presence of significant inter-cluster connections within hard graphs, which can confuse GNN methods that rely heavily on graph structures, potentially causing erroneous message propagation and biased clustering outcomes.
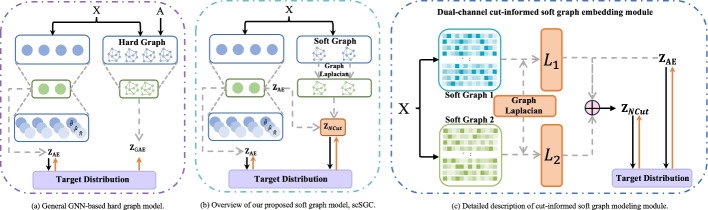
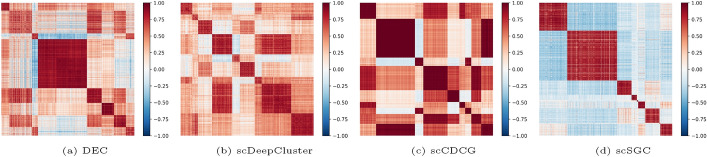
Results: To tackle these challenges, we introduce scSGC, a Soft Graph Clustering for single-cell RNA sequencing data, which aims to more accurately characterize continuous similarities among cells through non-binary edge weights, thereby mitigating the limitations of rigid data structures. The scSGC framework comprises three core components: (i) a zero-inflated negative binomial (ZINB)-based feature autoencoder designed to effectively handle the sparsity and dropout issues in scRNA-seq data; (ii) a dual-channel cut-informed soft graph embedding module, constructed through deep graph-cut information, capturing continuous similarities between cells while preserving the intrinsic data structures of scRNA-seq; and (iii) an optimal transport-based clustering optimization module, achieving optimal delineation of cell populations while maintaining high biological relevance.
Conclusion: By integrating dual-channel cut-informed soft graph representation learning, a ZINB-based feature autoencoder, and optimal transport-driven clustering optimization, scSGC effectively overcomes the challenges associated with traditional hard graph constructions in GNN methods. Extensive experiments across ten datasets demonstrate that scSGC outperforms 13 state-of-the-art clustering models in clustering accuracy, cell type annotation, and computational efficiency. These results highlight its substantial potential to advance scRNA-seq data analysis and deepen our understanding of cellular heterogeneity.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


