{"title":"具有染色体水平通用性的人类m6A修饰的混合表示学习。","authors":"Muhammad Tahir, Sheela Ramanna, Qian Liu","doi":"10.1093/bioadv/vbaf170","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong><math> <mrow> <mrow> <msup><mrow><mi>N</mi></mrow> <mn>6</mn></msup> </mrow> <mo>-</mo> <mtext>methyladenosine</mtext></mrow> </math> ( <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> ) is the most abundant internal modification in eukaryotic mRNA and plays essential roles in post-transcriptional gene regulation. While several deep learning approaches have been proposed to predict <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> sites, most suffer from limited chromosome-level generalizability due to evaluation on randomly split datasets.</p><p><strong>Results: </strong>In this study, we propose two novel hybrid deep learning models-Hybrid Model and Hybrid Deep Model-that integrate local sequence features (<i>k</i>-mers) and contextual embeddings via convolutional neural networks to improve predictive performance and generalization. We evaluate these models using both a Random-Split strategy and a more biologically realistic Leave-One-Chromosome-Out setting to ensure robustness across genomic regions. Our proposed models outperform the state-of-the-art m6A-TCPred model across all key evaluation metrics. Hybrid Deep Model achieves the highest accuracy under Random-Split, while Hybrid Model demonstrates superior generalization under Leave-One-Chromosome-Out, indicating that deep global representations may overfit in chromosome-independent settings. These findings underscore the importance of rigorous validation strategies and offer insights into designing robust <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> predictors.</p><p><strong>Availability and implementation: </strong>Source code and datasets are available at: https://github.com/malikmtahir/LOCO-m6A.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf170"},"PeriodicalIF":2.8000,"publicationDate":"2025-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288952/pdf/","citationCount":"0","resultStr":"{\"title\":\"Hybrid representation learning for human m<sup>6</sup>A modifications with chromosome-level generalizability.\",\"authors\":\"Muhammad Tahir, Sheela Ramanna, Qian Liu\",\"doi\":\"10.1093/bioadv/vbaf170\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong><math> <mrow> <mrow> <msup><mrow><mi>N</mi></mrow> <mn>6</mn></msup> </mrow> <mo>-</mo> <mtext>methyladenosine</mtext></mrow> </math> ( <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> ) is the most abundant internal modification in eukaryotic mRNA and plays essential roles in post-transcriptional gene regulation. While several deep learning approaches have been proposed to predict <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> sites, most suffer from limited chromosome-level generalizability due to evaluation on randomly split datasets.</p><p><strong>Results: </strong>In this study, we propose two novel hybrid deep learning models-Hybrid Model and Hybrid Deep Model-that integrate local sequence features (<i>k</i>-mers) and contextual embeddings via convolutional neural networks to improve predictive performance and generalization. We evaluate these models using both a Random-Split strategy and a more biologically realistic Leave-One-Chromosome-Out setting to ensure robustness across genomic regions. Our proposed models outperform the state-of-the-art m6A-TCPred model across all key evaluation metrics. Hybrid Deep Model achieves the highest accuracy under Random-Split, while Hybrid Model demonstrates superior generalization under Leave-One-Chromosome-Out, indicating that deep global representations may overfit in chromosome-independent settings. These findings underscore the importance of rigorous validation strategies and offer insights into designing robust <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> predictors.</p><p><strong>Availability and implementation: </strong>Source code and datasets are available at: https://github.com/malikmtahir/LOCO-m6A.</p>\",\"PeriodicalId\":72368,\"journal\":{\"name\":\"Bioinformatics advances\",\"volume\":\"5 1\",\"pages\":\"vbaf170\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288952/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioadv/vbaf170\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf170","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
动机:n6 -甲基腺苷(n6 - methylladenosine, m6 A)是真核生物mRNA中最丰富的内部修饰,在转录后基因调控中起重要作用。虽然已经提出了几种深度学习方法来预测m6a位点,但由于对随机分割的数据集进行评估,大多数方法在染色体水平上的泛化性有限。结果:在这项研究中,我们提出了两种新的混合深度学习模型——混合模型和混合深度模型,它们通过卷积神经网络集成了局部序列特征(k-mers)和上下文嵌入,以提高预测性能和泛化。我们使用随机分裂策略和更符合生物学现实的留一条染色体设置来评估这些模型,以确保跨基因组区域的稳健性。我们提出的模型在所有关键评估指标上都优于最先进的m6A-TCPred模型。Hybrid深度模型在Random-Split下达到最高的精度,而Hybrid模型在leave - one - chromosomeout下表现出更好的泛化,这表明深度全局表示在染色体无关的情况下可能会过拟合。这些发现强调了严格验证策略的重要性,并为设计稳健的m6a预测器提供了见解。可用性和实现:源代码和数据集可在:https://github.com/malikmtahir/LOCO-m6A。
Hybrid representation learning for human m6A modifications with chromosome-level generalizability.
Motivation: ( ) is the most abundant internal modification in eukaryotic mRNA and plays essential roles in post-transcriptional gene regulation. While several deep learning approaches have been proposed to predict sites, most suffer from limited chromosome-level generalizability due to evaluation on randomly split datasets.
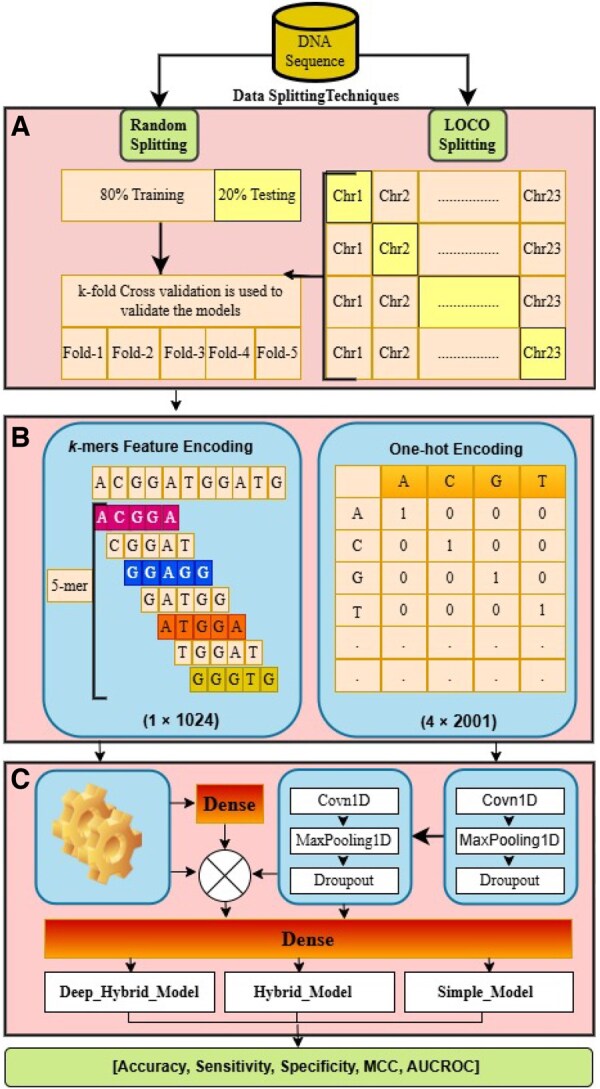
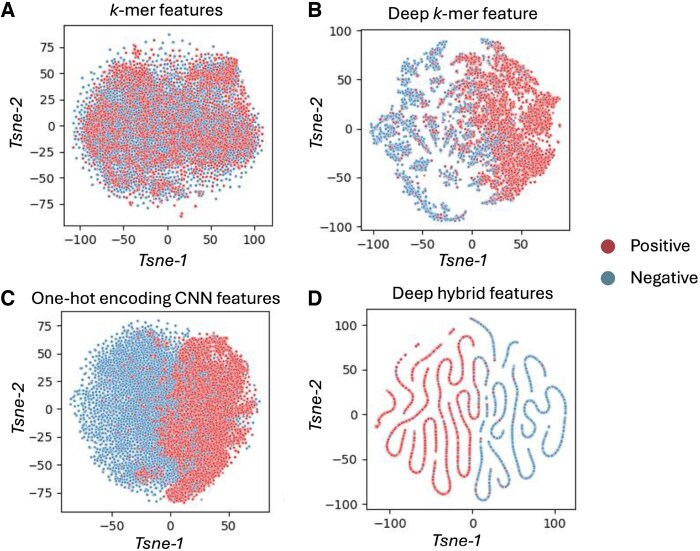
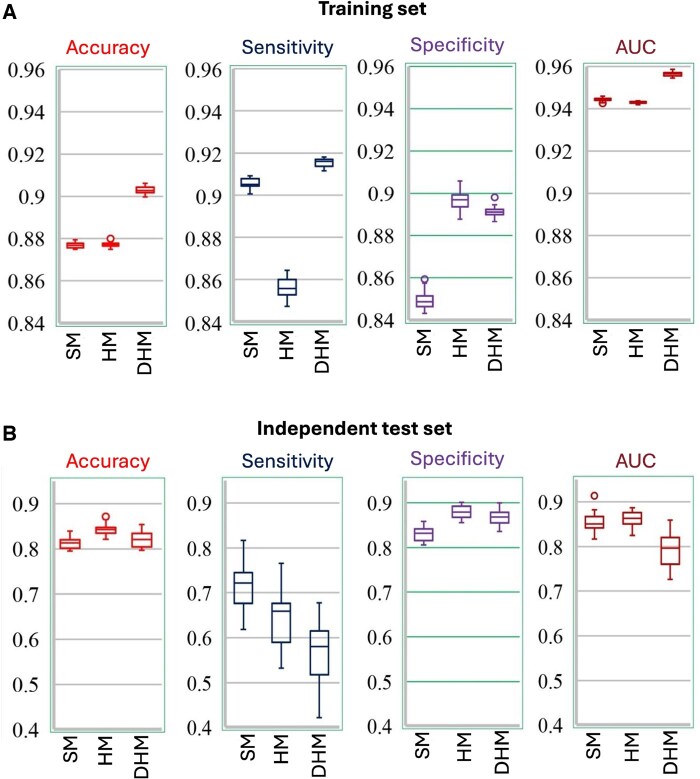
Results: In this study, we propose two novel hybrid deep learning models-Hybrid Model and Hybrid Deep Model-that integrate local sequence features (k-mers) and contextual embeddings via convolutional neural networks to improve predictive performance and generalization. We evaluate these models using both a Random-Split strategy and a more biologically realistic Leave-One-Chromosome-Out setting to ensure robustness across genomic regions. Our proposed models outperform the state-of-the-art m6A-TCPred model across all key evaluation metrics. Hybrid Deep Model achieves the highest accuracy under Random-Split, while Hybrid Model demonstrates superior generalization under Leave-One-Chromosome-Out, indicating that deep global representations may overfit in chromosome-independent settings. These findings underscore the importance of rigorous validation strategies and offer insights into designing robust predictors.
Availability and implementation: Source code and datasets are available at: https://github.com/malikmtahir/LOCO-m6A.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


