{"title":"abfold:更容易运行和比较AlphaFold 3, Boltz-1和Chai-1。","authors":"Luc G Elliott, Adam J Simpkin, Daniel J Rigden","doi":"10.1093/bioadv/vbaf153","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The latest generation of deep learning-based structure prediction methods enable accurate modelling of most proteins and many complexes. However, preparing inputs for the locally installed software is not always straightforward, and the results of local runs are not always presented in an ideally accessible fashion. Furthermore, it is not yet clear whether the latest tools perform equivalently for all types of target.</p><p><strong>Results: </strong>ABCFold facilitates the use of AlphaFold 3, Boltz-1, and Chai-1 with a standardized input to predict atomic structures, with Boltz-1 and Chai-1 being installed on runtime (if required). MSAs can be generated internally using either the JackHMMER MSA search within AlphaFold 3, or with the MMseqs2 API. Alternatively, users can provide their own custom MSAs. This therefore allows AlphaFold 3 to be installed and run without downloading the large databases needed for JackHMMER. There are also straightforward options to use templates, including custom templates. Results from all packages are treated in a unified fashion, enabling easy comparison of results from different methods. A variety of visualization options are available which include information on steric clashes.</p><p><strong>Availability and implementation: </strong>ABCFold is coded in Python and JavaScript. All scripts and associated documentation are available from https://github.com/rigdenlab/ABCFold or https://pypi.org/project/ABCFold/.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf153"},"PeriodicalIF":2.8000,"publicationDate":"2025-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12287924/pdf/","citationCount":"0","resultStr":"{\"title\":\"ABCFold: easier running and comparison of AlphaFold 3, Boltz-1, and Chai-1.\",\"authors\":\"Luc G Elliott, Adam J Simpkin, Daniel J Rigden\",\"doi\":\"10.1093/bioadv/vbaf153\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>The latest generation of deep learning-based structure prediction methods enable accurate modelling of most proteins and many complexes. However, preparing inputs for the locally installed software is not always straightforward, and the results of local runs are not always presented in an ideally accessible fashion. Furthermore, it is not yet clear whether the latest tools perform equivalently for all types of target.</p><p><strong>Results: </strong>ABCFold facilitates the use of AlphaFold 3, Boltz-1, and Chai-1 with a standardized input to predict atomic structures, with Boltz-1 and Chai-1 being installed on runtime (if required). MSAs can be generated internally using either the JackHMMER MSA search within AlphaFold 3, or with the MMseqs2 API. Alternatively, users can provide their own custom MSAs. This therefore allows AlphaFold 3 to be installed and run without downloading the large databases needed for JackHMMER. There are also straightforward options to use templates, including custom templates. Results from all packages are treated in a unified fashion, enabling easy comparison of results from different methods. A variety of visualization options are available which include information on steric clashes.</p><p><strong>Availability and implementation: </strong>ABCFold is coded in Python and JavaScript. All scripts and associated documentation are available from https://github.com/rigdenlab/ABCFold or https://pypi.org/project/ABCFold/.</p>\",\"PeriodicalId\":72368,\"journal\":{\"name\":\"Bioinformatics advances\",\"volume\":\"5 1\",\"pages\":\"vbaf153\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12287924/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioadv/vbaf153\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf153","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
ABCFold: easier running and comparison of AlphaFold 3, Boltz-1, and Chai-1.
Motivation: The latest generation of deep learning-based structure prediction methods enable accurate modelling of most proteins and many complexes. However, preparing inputs for the locally installed software is not always straightforward, and the results of local runs are not always presented in an ideally accessible fashion. Furthermore, it is not yet clear whether the latest tools perform equivalently for all types of target.
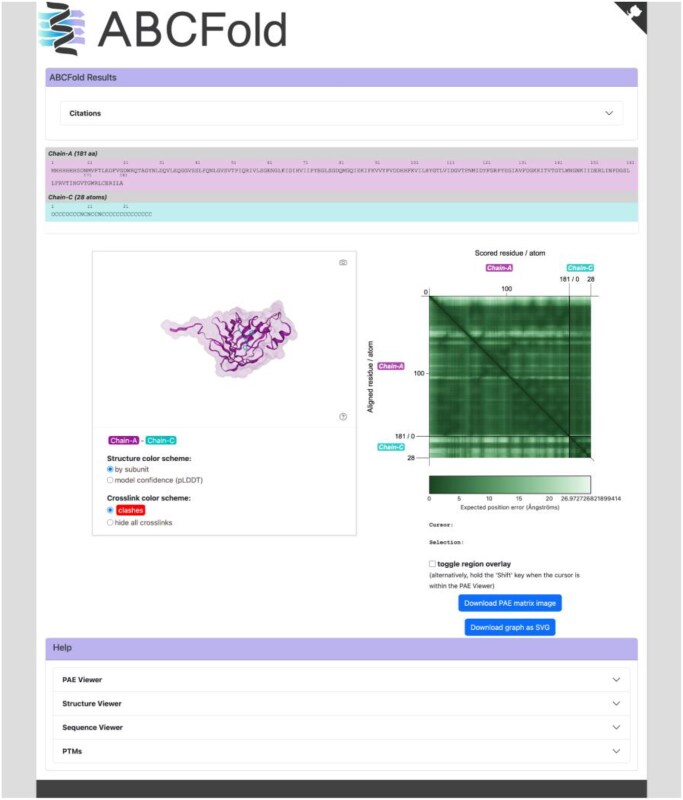
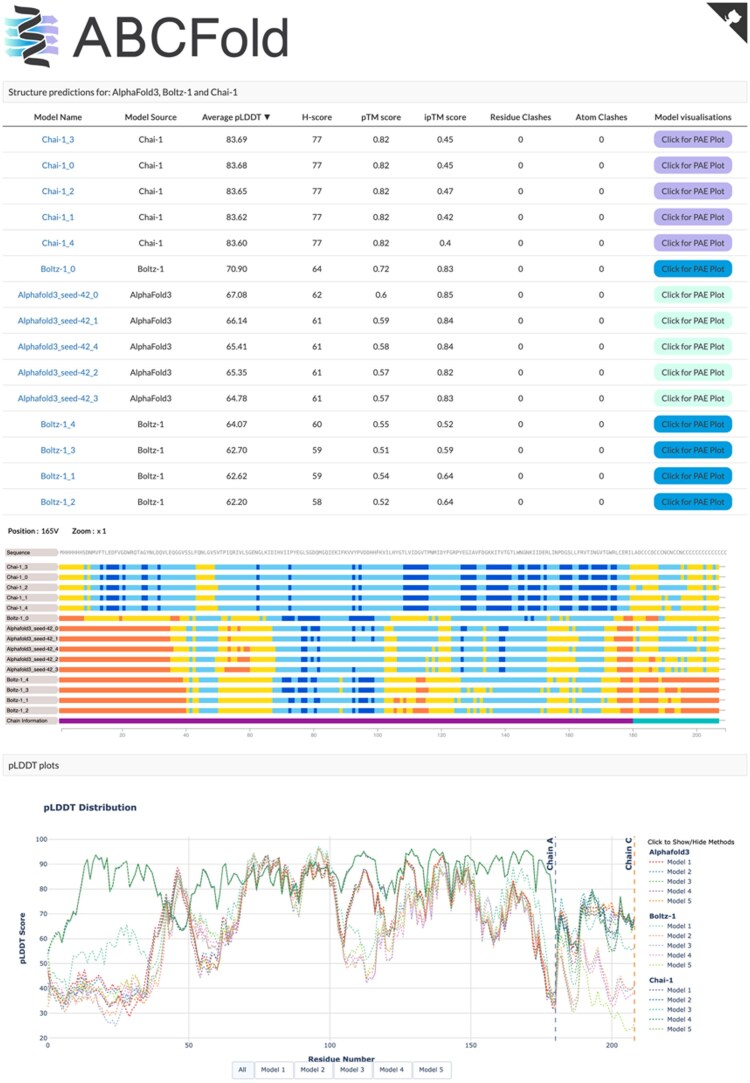
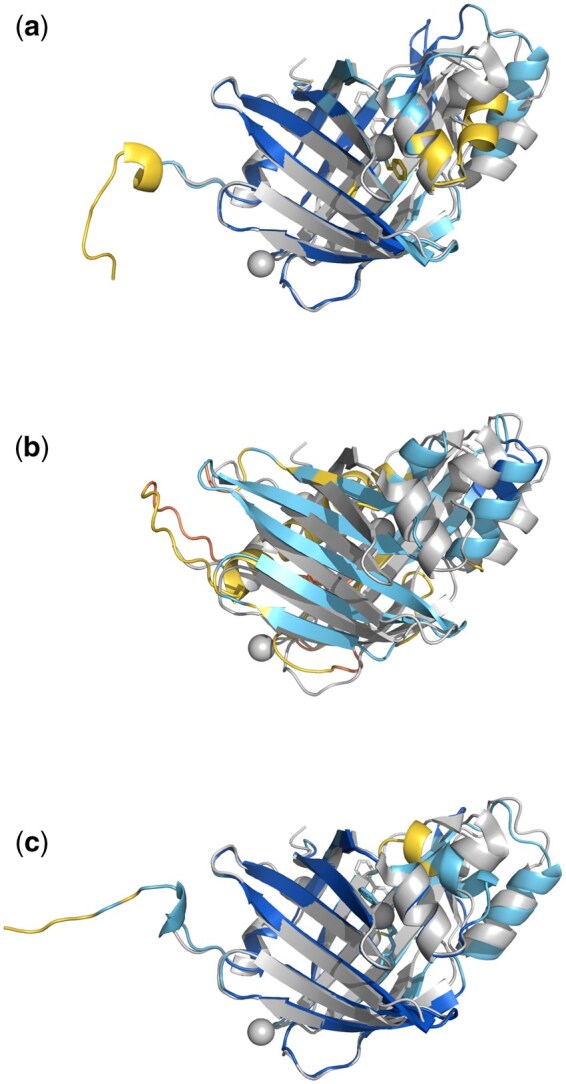
Results: ABCFold facilitates the use of AlphaFold 3, Boltz-1, and Chai-1 with a standardized input to predict atomic structures, with Boltz-1 and Chai-1 being installed on runtime (if required). MSAs can be generated internally using either the JackHMMER MSA search within AlphaFold 3, or with the MMseqs2 API. Alternatively, users can provide their own custom MSAs. This therefore allows AlphaFold 3 to be installed and run without downloading the large databases needed for JackHMMER. There are also straightforward options to use templates, including custom templates. Results from all packages are treated in a unified fashion, enabling easy comparison of results from different methods. A variety of visualization options are available which include information on steric clashes.
Availability and implementation: ABCFold is coded in Python and JavaScript. All scripts and associated documentation are available from https://github.com/rigdenlab/ABCFold or https://pypi.org/project/ABCFold/.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


