María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos
{"title":"Tay-Sachs病的诊断、病理机制、临床影响和未来治疗前景的进展。","authors":"María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos","doi":"10.3390/neurolint17070098","DOIUrl":null,"url":null,"abstract":"<p><p>Tay-Sachs disease (TSD) is a rare and severe neurodegenerative disorder inherited in an autosomal recessive manner. It is caused by a deficiency of the enzyme hexosaminidase A, which is responsible for the degradation of GM2 gangliosides-lipids that accumulate in the nerve cells of the central nervous system. The inability to break down these lipids leads to their progressive accumulation, resulting in irreversible brain damage. Mechanistically, TSD is caused by mutations in the <i>HEXA</i> gene, which encodes the alpha subunit of hexosaminidase A. These mutations disrupt enzyme activity and alter cellular pathways involved in lysosomal lipid degradation. Although Tay-Sachs specifically involves the alpha subunit, similar clinical features can be seen in Sandhoff disease, a related disorder caused by mutations in the <i>HEXB</i> gene, which encodes the beta subunit shared by hexosaminidase A and B. Tay-Sachs is classified into three clinical forms according to age of onset and symptom severity: the classic infantile form, which is the most common and severe; a juvenile (subacute) form; and an adult-onset form, which progresses more slowly and tends to present with milder symptoms. Diagnosis is based on enzymatic testing showing reduced or absent hexosaminidase A activity, confirmed by genetic testing. Prenatal diagnosis and genetic counseling play a key role in prevention and reproductive decision-making, especially in high-risk populations. Although no curative treatment currently exists, ongoing research is exploring gene therapy, enzyme replacement, and pharmacological approaches. Certain compounds, such as gemfibrozil, have shown potential to slow symptom progression. Early diagnosis and multidisciplinary care are essential to improving quality of life, although therapeutic options remain limited due to the progressive nature of the disease.</p>","PeriodicalId":19130,"journal":{"name":"Neurology International","volume":"17 7","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2025-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12298450/pdf/","citationCount":"0","resultStr":"{\"title\":\"Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay-Sachs Disease.\",\"authors\":\"María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos\",\"doi\":\"10.3390/neurolint17070098\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Tay-Sachs disease (TSD) is a rare and severe neurodegenerative disorder inherited in an autosomal recessive manner. It is caused by a deficiency of the enzyme hexosaminidase A, which is responsible for the degradation of GM2 gangliosides-lipids that accumulate in the nerve cells of the central nervous system. The inability to break down these lipids leads to their progressive accumulation, resulting in irreversible brain damage. Mechanistically, TSD is caused by mutations in the <i>HEXA</i> gene, which encodes the alpha subunit of hexosaminidase A. These mutations disrupt enzyme activity and alter cellular pathways involved in lysosomal lipid degradation. Although Tay-Sachs specifically involves the alpha subunit, similar clinical features can be seen in Sandhoff disease, a related disorder caused by mutations in the <i>HEXB</i> gene, which encodes the beta subunit shared by hexosaminidase A and B. Tay-Sachs is classified into three clinical forms according to age of onset and symptom severity: the classic infantile form, which is the most common and severe; a juvenile (subacute) form; and an adult-onset form, which progresses more slowly and tends to present with milder symptoms. Diagnosis is based on enzymatic testing showing reduced or absent hexosaminidase A activity, confirmed by genetic testing. Prenatal diagnosis and genetic counseling play a key role in prevention and reproductive decision-making, especially in high-risk populations. Although no curative treatment currently exists, ongoing research is exploring gene therapy, enzyme replacement, and pharmacological approaches. Certain compounds, such as gemfibrozil, have shown potential to slow symptom progression. Early diagnosis and multidisciplinary care are essential to improving quality of life, although therapeutic options remain limited due to the progressive nature of the disease.</p>\",\"PeriodicalId\":19130,\"journal\":{\"name\":\"Neurology International\",\"volume\":\"17 7\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2025-06-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12298450/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology International\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/neurolint17070098\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology International","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/neurolint17070098","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay-Sachs Disease.
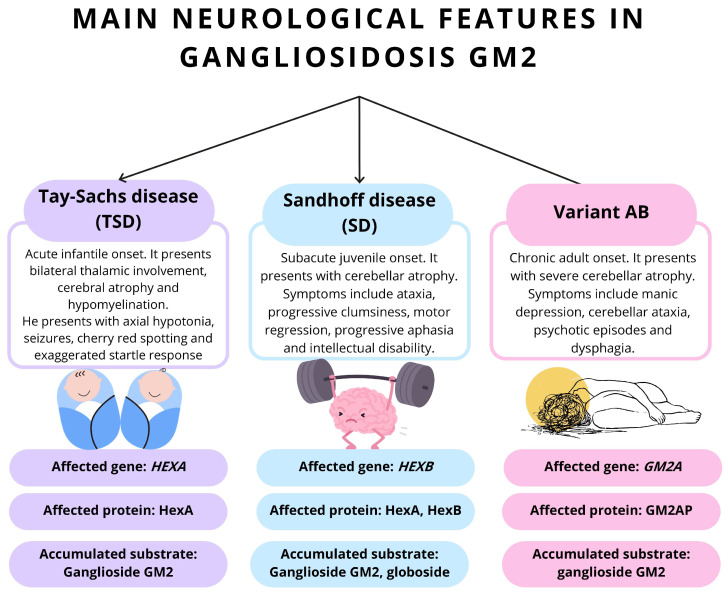
Tay-Sachs disease (TSD) is a rare and severe neurodegenerative disorder inherited in an autosomal recessive manner. It is caused by a deficiency of the enzyme hexosaminidase A, which is responsible for the degradation of GM2 gangliosides-lipids that accumulate in the nerve cells of the central nervous system. The inability to break down these lipids leads to their progressive accumulation, resulting in irreversible brain damage. Mechanistically, TSD is caused by mutations in the HEXA gene, which encodes the alpha subunit of hexosaminidase A. These mutations disrupt enzyme activity and alter cellular pathways involved in lysosomal lipid degradation. Although Tay-Sachs specifically involves the alpha subunit, similar clinical features can be seen in Sandhoff disease, a related disorder caused by mutations in the HEXB gene, which encodes the beta subunit shared by hexosaminidase A and B. Tay-Sachs is classified into three clinical forms according to age of onset and symptom severity: the classic infantile form, which is the most common and severe; a juvenile (subacute) form; and an adult-onset form, which progresses more slowly and tends to present with milder symptoms. Diagnosis is based on enzymatic testing showing reduced or absent hexosaminidase A activity, confirmed by genetic testing. Prenatal diagnosis and genetic counseling play a key role in prevention and reproductive decision-making, especially in high-risk populations. Although no curative treatment currently exists, ongoing research is exploring gene therapy, enzyme replacement, and pharmacological approaches. Certain compounds, such as gemfibrozil, have shown potential to slow symptom progression. Early diagnosis and multidisciplinary care are essential to improving quality of life, although therapeutic options remain limited due to the progressive nature of the disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


