通过分级分子表征和细胞系潜伏空间融合预测协同药物组合
IF 4.8
2区 医学
Q1 COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS
引用次数: 0
摘要
癌症治疗往往受益于药物组合的协同效应。预测这些协同作用对于开发有效的联合疗法至关重要。现有的深度学习模型通常使用单个图结构来表示药物,并直接使用细胞系基因表达谱,这可能导致详细分子特征的丢失和噪声的引入。本研究旨在通过开发一种新的深度学习模型来改善药物协同作用的预测,该模型可以在多个水平上捕获药物分子的局部和全局特征,并减少细胞系数据中的噪声。该模型在节点级、基序级和图级上分层表示药物分子,以获取全面的特征信息。利用Mamba模块和基于图注意的卷积有效地提取药物对的深度特征信息。编码器-解码器结构将细胞系投射到潜在空间,通过星形操作和注意机制将噪声降至最低,并增强与药物对数据的整合。该模型在包含来自各种癌细胞系的药物反应数据的基准数据集上进行了训练和验证。针对基准数据集的模型评估显示,与现有方法相比,该模型具有优越的性能。这些结果表明,该模型可以更准确地预测协同抗癌药物组合,为联合治疗的设计提供可靠的支持。增强的预测准确性可以促进有效药物组合的发现,潜在地加速个性化癌症治疗的发展。本文章由计算机程序翻译,如有差异,请以英文原文为准。
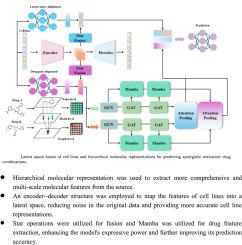
Predicting synergistic drug combinations via hierarchical molecular representation and cell line latent space fusion
Cancer treatment often benefits from the synergistic effects of drug combinations. Predicting these synergies is critical for developing effective combination therapies. Existing deep learning models typically represent drugs using a single graph structure and use cell line gene expression profiles directly, potentially leading to loss of detailed molecular features and introduction of noise. This study aims to improve the prediction of drug synergy by developing a novel deep learning model that captures both local and global features of drug molecules at multiple levels and reduces noise in cell line data. The proposed model hierarchically represents drug molecules at the node, motif, and graph levels to capture comprehensive feature information. The Mamba module and graph attention-based convolution are employed to effectively extract deep feature information from drug pairs. An encoder–decoder structure projects cell lines into a latent space, minimizing noise and enhancing the integration with drug pair data through star operations and an attention mechanism. The model was trained and validated on benchmark datasets containing drug response data from various cancer cell lines. The evaluation of the model against benchmark datasets demonstrated superior performance compared to existing methods. These results indicate that the model can more accurately predict synergistic anticancer drug combinations, providing reliable support for the design of combination therapies. The enhanced predictive accuracy can facilitate the discovery of effective drug combinations, potentially accelerating the development of personalized cancer treatments.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computer methods and programs in biomedicine
工程技术-工程:生物医学
CiteScore
12.30
自引率
6.60%
发文量
601
审稿时长
135 days
期刊介绍:
To encourage the development of formal computing methods, and their application in biomedical research and medical practice, by illustration of fundamental principles in biomedical informatics research; to stimulate basic research into application software design; to report the state of research of biomedical information processing projects; to report new computer methodologies applied in biomedical areas; the eventual distribution of demonstrable software to avoid duplication of effort; to provide a forum for discussion and improvement of existing software; to optimize contact between national organizations and regional user groups by promoting an international exchange of information on formal methods, standards and software in biomedicine.
Computer Methods and Programs in Biomedicine covers computing methodology and software systems derived from computing science for implementation in all aspects of biomedical research and medical practice. It is designed to serve: biochemists; biologists; geneticists; immunologists; neuroscientists; pharmacologists; toxicologists; clinicians; epidemiologists; psychiatrists; psychologists; cardiologists; chemists; (radio)physicists; computer scientists; programmers and systems analysts; biomedical, clinical, electrical and other engineers; teachers of medical informatics and users of educational software.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


