Amit Kumar Pradhan, Athul Sudheendranath, Jitesh Arora, Rahul Dahiya and Sajesh P. Thomas
{"title":"高Z′多态性是亚稳态的吗?从药物多态性的见解","authors":"Amit Kumar Pradhan, Athul Sudheendranath, Jitesh Arora, Rahul Dahiya and Sajesh P. Thomas","doi":"10.1039/D5CP02122G","DOIUrl":null,"url":null,"abstract":"<p >High-<em>Z</em>′ polymorphs are hypothesized to be kinetically trapped products and hence less stable than their low-<em>Z</em>′ counterparts (<em>Z</em>′ = number of symmetry-independent molecules in a crystal structure). Although the existence of such a symmetry–stability relationship would be of fundamental importance to the structural chemistry of molecular crystals, it has yet to be supported by experimental or computational evidence. A major challenge in analyzing high-<em>Z</em>′ crystal structures is the large number of atoms, which makes accurate quantum chemical evaluation of their lattice cohesive energies (LCEs) computationally intensive. A systematic test of this hypothesis in drug polymorphs is made feasible by the CE-B3LYP method, which uses pairwise summation of interaction energies for LCE estimation. Here, we have analyzed 15 drugs (49 polymorphs) of low- and high-<em>Z</em>′ crystal forms in terms of lattice cohesive energies, Kitaigorodskii packing indices (KPI), in-crystal molecular volumes, and crystal densities. Our results show no direct relation between <em>Z</em>′ and the stability of drug polymorphs.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 34","pages":" 17779-17786"},"PeriodicalIF":2.9000,"publicationDate":"2025-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Are high-Z′ polymorphs metastable? Insight from pharmaceutical polymorphs\",\"authors\":\"Amit Kumar Pradhan, Athul Sudheendranath, Jitesh Arora, Rahul Dahiya and Sajesh P. Thomas\",\"doi\":\"10.1039/D5CP02122G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >High-<em>Z</em>′ polymorphs are hypothesized to be kinetically trapped products and hence less stable than their low-<em>Z</em>′ counterparts (<em>Z</em>′ = number of symmetry-independent molecules in a crystal structure). Although the existence of such a symmetry–stability relationship would be of fundamental importance to the structural chemistry of molecular crystals, it has yet to be supported by experimental or computational evidence. A major challenge in analyzing high-<em>Z</em>′ crystal structures is the large number of atoms, which makes accurate quantum chemical evaluation of their lattice cohesive energies (LCEs) computationally intensive. A systematic test of this hypothesis in drug polymorphs is made feasible by the CE-B3LYP method, which uses pairwise summation of interaction energies for LCE estimation. Here, we have analyzed 15 drugs (49 polymorphs) of low- and high-<em>Z</em>′ crystal forms in terms of lattice cohesive energies, Kitaigorodskii packing indices (KPI), in-crystal molecular volumes, and crystal densities. Our results show no direct relation between <em>Z</em>′ and the stability of drug polymorphs.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 34\",\"pages\":\" 17779-17786\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02122g\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02122g","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Are high-Z′ polymorphs metastable? Insight from pharmaceutical polymorphs
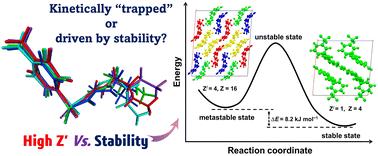
High-Z′ polymorphs are hypothesized to be kinetically trapped products and hence less stable than their low-Z′ counterparts (Z′ = number of symmetry-independent molecules in a crystal structure). Although the existence of such a symmetry–stability relationship would be of fundamental importance to the structural chemistry of molecular crystals, it has yet to be supported by experimental or computational evidence. A major challenge in analyzing high-Z′ crystal structures is the large number of atoms, which makes accurate quantum chemical evaluation of their lattice cohesive energies (LCEs) computationally intensive. A systematic test of this hypothesis in drug polymorphs is made feasible by the CE-B3LYP method, which uses pairwise summation of interaction energies for LCE estimation. Here, we have analyzed 15 drugs (49 polymorphs) of low- and high-Z′ crystal forms in terms of lattice cohesive energies, Kitaigorodskii packing indices (KPI), in-crystal molecular volumes, and crystal densities. Our results show no direct relation between Z′ and the stability of drug polymorphs.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


