Franz Leonard Böge, Helena U Zacharias, Stefanie C Becker, Klaus Jung
{"title":"基于mRNA数据,利用深度神经网络和LASSO回归预测miRNA表达变化。","authors":"Franz Leonard Böge, Helena U Zacharias, Stefanie C Becker, Klaus Jung","doi":"10.3389/fbinf.2025.1566162","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Since the rise of molecular high-throughput technologies, many diseases are now studied on multiple omics layers in parallel. Understanding the interplay between microRNAs (miRNA) and their target mRNAs is important to understand the molecular level of diseases. While much public data from mRNA experiments are available for many diseases, few paired datasets with both miRNA and mRNA expression profiles are available. This study aimed to assess the possibility of predicting miRNA expression data based on mRNA expression data, serving as a proof of principle that such cross-omics predictions are feasible. Furthermore, current research relies on target databases where information about miRNA-target relationships is provided based on experimental and computational studies.</p><p><strong>Methods: </strong>To make use of publicly available mRNA profiles, we investigate the ability of artificial deep neural networks and linear least absolute shrinkage and selection operator (LASSO) regression to predict unknown miRNA expression profiles. We evaluate the approach using seven paired miRNA/mRNA expression datasets, four from studies on West Nile virus infection in mouse tissues and three from human immunodeficiency virus (HIV) infection in human tissues. We assessed the performance of each model first by within-data evaluations and second by cross-study evaluations. Furthermore, we investigated whether data augmentation or separate models for data from diseased and non-diseased samples can improve the prediction performance.</p><p><strong>Results: </strong>In general, most settings achieved strong correlations at the Level of individual samples. In some datasets and settings, correlations of log-fold changes and p-values from differential expression analysis (DEA) between true and predicted miRNA profiles can be observed. Correlation between log fold changes could also be seen in a cross-study evaluation for the HIV datasets. Data augmentation consistently improved performance in neural networks, while its impact on LASSO models was not significant.</p><p><strong>Discussion: </strong>Overall, cross-omics prediction of expression profiles appears possible, even with some correlations on the Level of the differential expression analysis.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"5 ","pages":"1566162"},"PeriodicalIF":3.9000,"publicationDate":"2025-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12279838/pdf/","citationCount":"0","resultStr":"{\"title\":\"Using deep neural networks and LASSO regression to predict miRNA expression changes based on mRNA data.\",\"authors\":\"Franz Leonard Böge, Helena U Zacharias, Stefanie C Becker, Klaus Jung\",\"doi\":\"10.3389/fbinf.2025.1566162\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Since the rise of molecular high-throughput technologies, many diseases are now studied on multiple omics layers in parallel. Understanding the interplay between microRNAs (miRNA) and their target mRNAs is important to understand the molecular level of diseases. While much public data from mRNA experiments are available for many diseases, few paired datasets with both miRNA and mRNA expression profiles are available. This study aimed to assess the possibility of predicting miRNA expression data based on mRNA expression data, serving as a proof of principle that such cross-omics predictions are feasible. Furthermore, current research relies on target databases where information about miRNA-target relationships is provided based on experimental and computational studies.</p><p><strong>Methods: </strong>To make use of publicly available mRNA profiles, we investigate the ability of artificial deep neural networks and linear least absolute shrinkage and selection operator (LASSO) regression to predict unknown miRNA expression profiles. We evaluate the approach using seven paired miRNA/mRNA expression datasets, four from studies on West Nile virus infection in mouse tissues and three from human immunodeficiency virus (HIV) infection in human tissues. We assessed the performance of each model first by within-data evaluations and second by cross-study evaluations. Furthermore, we investigated whether data augmentation or separate models for data from diseased and non-diseased samples can improve the prediction performance.</p><p><strong>Results: </strong>In general, most settings achieved strong correlations at the Level of individual samples. In some datasets and settings, correlations of log-fold changes and p-values from differential expression analysis (DEA) between true and predicted miRNA profiles can be observed. Correlation between log fold changes could also be seen in a cross-study evaluation for the HIV datasets. Data augmentation consistently improved performance in neural networks, while its impact on LASSO models was not significant.</p><p><strong>Discussion: </strong>Overall, cross-omics prediction of expression profiles appears possible, even with some correlations on the Level of the differential expression analysis.</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"5 \",\"pages\":\"1566162\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2025-07-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12279838/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2025.1566162\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2025.1566162","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Using deep neural networks and LASSO regression to predict miRNA expression changes based on mRNA data.
Introduction: Since the rise of molecular high-throughput technologies, many diseases are now studied on multiple omics layers in parallel. Understanding the interplay between microRNAs (miRNA) and their target mRNAs is important to understand the molecular level of diseases. While much public data from mRNA experiments are available for many diseases, few paired datasets with both miRNA and mRNA expression profiles are available. This study aimed to assess the possibility of predicting miRNA expression data based on mRNA expression data, serving as a proof of principle that such cross-omics predictions are feasible. Furthermore, current research relies on target databases where information about miRNA-target relationships is provided based on experimental and computational studies.
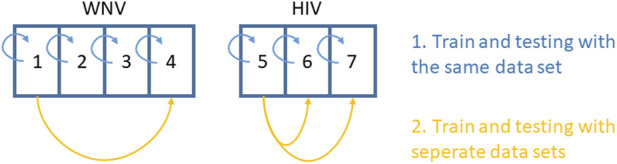
Methods: To make use of publicly available mRNA profiles, we investigate the ability of artificial deep neural networks and linear least absolute shrinkage and selection operator (LASSO) regression to predict unknown miRNA expression profiles. We evaluate the approach using seven paired miRNA/mRNA expression datasets, four from studies on West Nile virus infection in mouse tissues and three from human immunodeficiency virus (HIV) infection in human tissues. We assessed the performance of each model first by within-data evaluations and second by cross-study evaluations. Furthermore, we investigated whether data augmentation or separate models for data from diseased and non-diseased samples can improve the prediction performance.
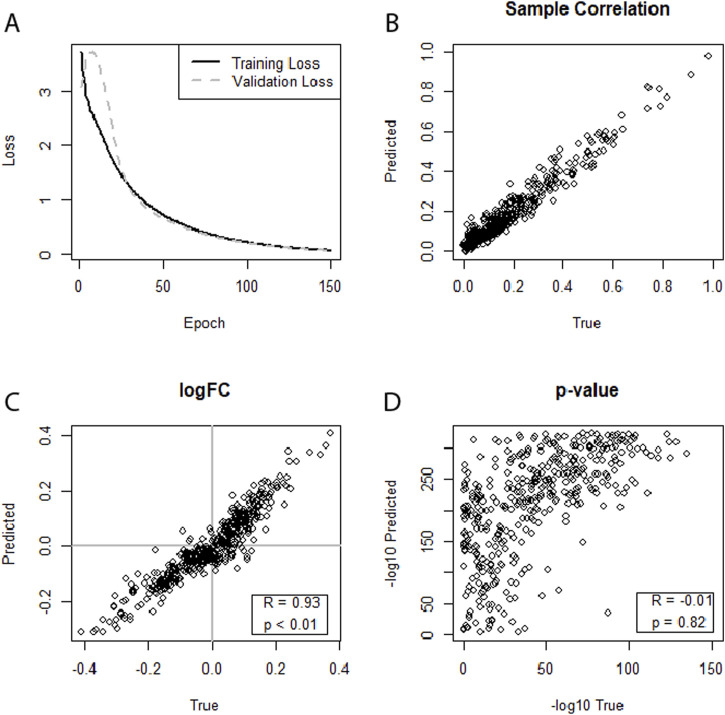
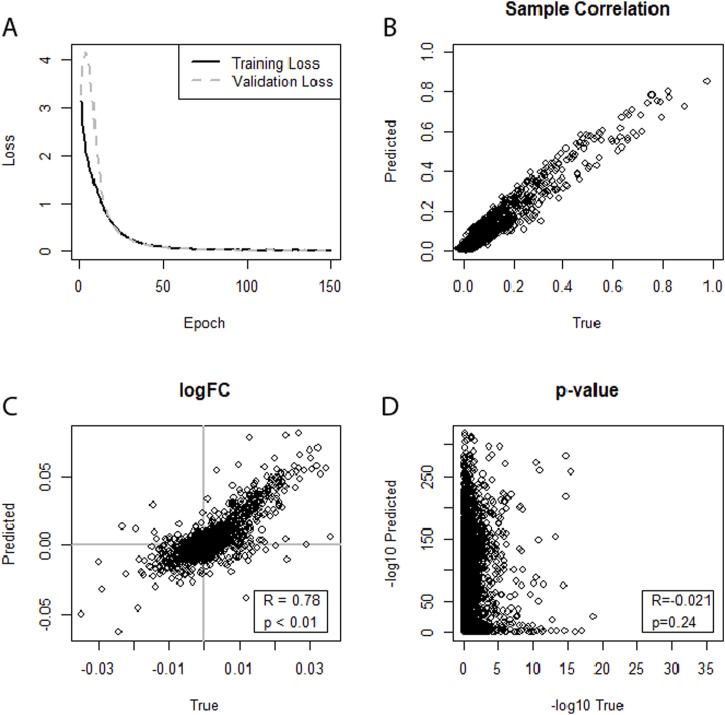
Results: In general, most settings achieved strong correlations at the Level of individual samples. In some datasets and settings, correlations of log-fold changes and p-values from differential expression analysis (DEA) between true and predicted miRNA profiles can be observed. Correlation between log fold changes could also be seen in a cross-study evaluation for the HIV datasets. Data augmentation consistently improved performance in neural networks, while its impact on LASSO models was not significant.
Discussion: Overall, cross-omics prediction of expression profiles appears possible, even with some correlations on the Level of the differential expression analysis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


