{"title":"V3-V4和FL-16S rRNA扩增子测序方法用于气管切开术吸入物微生物群落分析的评价","authors":"A Gupta, V S Cooper, A C Zemke","doi":"10.1128/msphere.00388-25","DOIUrl":null,"url":null,"abstract":"<p><p>Respiratory infections pose a significant risk for people requiring prolonged mechanical ventilation, yet limited information exists regarding the complex microbiome dynamics of people with tracheostomies during chronic critical illness. Oxford Nanopore Technologies (ONT) long-read sequencing allows for full-length 16S rRNA amplicon sequencing, providing enhanced species-level understanding of the respiratory microbiome. We validated ONT-based FL-16S amplicon sequencing for microbial insights from tracheal aspirates by comparing results with those of Illumina V3-V4 amplicon sequencing. Comparisons were made on a standardized microbial community and tracheal aspirates using multiple DNA extraction kits. Conventional short-read bioinformatic pipelines are suboptimal for processing longer, error-prone ONT reads. The Emu bioinformatics pipeline, specifically designed for ONT FL-16S reads, enhances the accuracy but necessitates validation for tracheal aspirates. In this study, we compared the analysis of FL-16S reads using Emu to the standardized V3-V4 read analysis with QIIME2. Our findings demonstrate that at the same sequencing read depth, FL-16S sequencing analysis with Emu yields comparable alpha and taxonomic diversity metrics, while providing superior species-level resolution compared to V3-V4 amplicon sequencing of tracheal aspirates. Our results show that tracheal aspirates during chronic critical illness are low-diversity samples, with most pathogenic genera represented by a single species. However, members of the oral microbiota <i>Streptococcus</i> and <i>Prevotella</i> are represented by multiple species.</p><p><strong>Importance: </strong>The role of the respiratory microbiome in shaping outcomes for patients with chronic critical illness undergoing prolonged mechanical ventilation via a tracheostomy remains poorly understood, despite its potential to drive infections and complicate recovery. Current methods, such as short-read 16S rRNA sequencing, lack taxonomic resolution to track pathogens at the species level, limiting clinical insights. Our study addresses this gap by validating ONT-based full-length (FL)-16S rRNA sequencing, a method that achieves species-level taxonomic precision critical for analyzing complex respiratory microbiomes. We benchmarked the microbiome composition of tracheal aspirates from ONT FL-16S rRNA workflows against Illumina V3-V4 data to demonstrate that long-read sequencing delivers comparable diversity profiles while resolving species-level diversity of clinically relevant species and microbes associated with the oral microbiome.</p>","PeriodicalId":19052,"journal":{"name":"mSphere","volume":" ","pages":"e0038825"},"PeriodicalIF":3.1000,"publicationDate":"2025-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12379586/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evaluation of V3-V4 and FL-16S rRNA amplicon sequencing approach for microbiota community analysis of tracheostomy aspirates.\",\"authors\":\"A Gupta, V S Cooper, A C Zemke\",\"doi\":\"10.1128/msphere.00388-25\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Respiratory infections pose a significant risk for people requiring prolonged mechanical ventilation, yet limited information exists regarding the complex microbiome dynamics of people with tracheostomies during chronic critical illness. Oxford Nanopore Technologies (ONT) long-read sequencing allows for full-length 16S rRNA amplicon sequencing, providing enhanced species-level understanding of the respiratory microbiome. We validated ONT-based FL-16S amplicon sequencing for microbial insights from tracheal aspirates by comparing results with those of Illumina V3-V4 amplicon sequencing. Comparisons were made on a standardized microbial community and tracheal aspirates using multiple DNA extraction kits. Conventional short-read bioinformatic pipelines are suboptimal for processing longer, error-prone ONT reads. The Emu bioinformatics pipeline, specifically designed for ONT FL-16S reads, enhances the accuracy but necessitates validation for tracheal aspirates. In this study, we compared the analysis of FL-16S reads using Emu to the standardized V3-V4 read analysis with QIIME2. Our findings demonstrate that at the same sequencing read depth, FL-16S sequencing analysis with Emu yields comparable alpha and taxonomic diversity metrics, while providing superior species-level resolution compared to V3-V4 amplicon sequencing of tracheal aspirates. Our results show that tracheal aspirates during chronic critical illness are low-diversity samples, with most pathogenic genera represented by a single species. However, members of the oral microbiota <i>Streptococcus</i> and <i>Prevotella</i> are represented by multiple species.</p><p><strong>Importance: </strong>The role of the respiratory microbiome in shaping outcomes for patients with chronic critical illness undergoing prolonged mechanical ventilation via a tracheostomy remains poorly understood, despite its potential to drive infections and complicate recovery. Current methods, such as short-read 16S rRNA sequencing, lack taxonomic resolution to track pathogens at the species level, limiting clinical insights. Our study addresses this gap by validating ONT-based full-length (FL)-16S rRNA sequencing, a method that achieves species-level taxonomic precision critical for analyzing complex respiratory microbiomes. We benchmarked the microbiome composition of tracheal aspirates from ONT FL-16S rRNA workflows against Illumina V3-V4 data to demonstrate that long-read sequencing delivers comparable diversity profiles while resolving species-level diversity of clinically relevant species and microbes associated with the oral microbiome.</p>\",\"PeriodicalId\":19052,\"journal\":{\"name\":\"mSphere\",\"volume\":\" \",\"pages\":\"e0038825\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2025-08-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12379586/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"mSphere\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1128/msphere.00388-25\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"mSphere","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1128/msphere.00388-25","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Evaluation of V3-V4 and FL-16S rRNA amplicon sequencing approach for microbiota community analysis of tracheostomy aspirates.
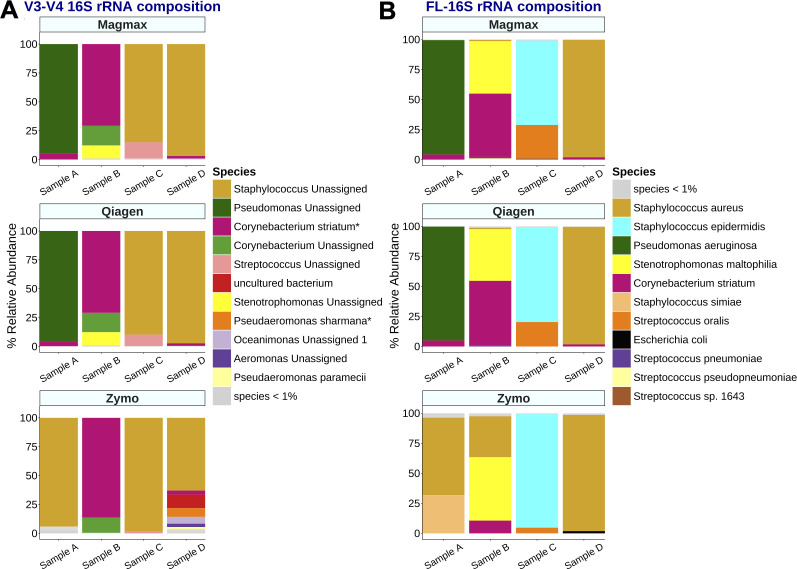
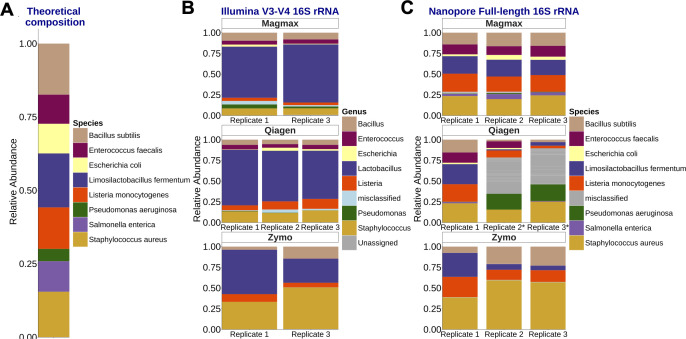
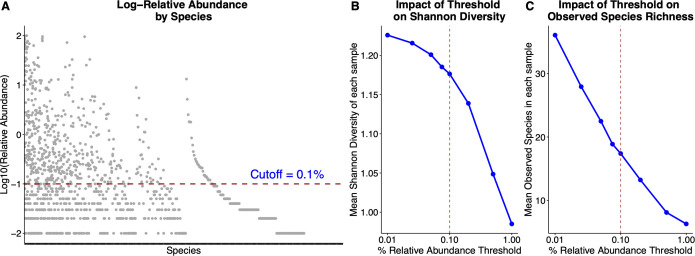
Respiratory infections pose a significant risk for people requiring prolonged mechanical ventilation, yet limited information exists regarding the complex microbiome dynamics of people with tracheostomies during chronic critical illness. Oxford Nanopore Technologies (ONT) long-read sequencing allows for full-length 16S rRNA amplicon sequencing, providing enhanced species-level understanding of the respiratory microbiome. We validated ONT-based FL-16S amplicon sequencing for microbial insights from tracheal aspirates by comparing results with those of Illumina V3-V4 amplicon sequencing. Comparisons were made on a standardized microbial community and tracheal aspirates using multiple DNA extraction kits. Conventional short-read bioinformatic pipelines are suboptimal for processing longer, error-prone ONT reads. The Emu bioinformatics pipeline, specifically designed for ONT FL-16S reads, enhances the accuracy but necessitates validation for tracheal aspirates. In this study, we compared the analysis of FL-16S reads using Emu to the standardized V3-V4 read analysis with QIIME2. Our findings demonstrate that at the same sequencing read depth, FL-16S sequencing analysis with Emu yields comparable alpha and taxonomic diversity metrics, while providing superior species-level resolution compared to V3-V4 amplicon sequencing of tracheal aspirates. Our results show that tracheal aspirates during chronic critical illness are low-diversity samples, with most pathogenic genera represented by a single species. However, members of the oral microbiota Streptococcus and Prevotella are represented by multiple species.
Importance: The role of the respiratory microbiome in shaping outcomes for patients with chronic critical illness undergoing prolonged mechanical ventilation via a tracheostomy remains poorly understood, despite its potential to drive infections and complicate recovery. Current methods, such as short-read 16S rRNA sequencing, lack taxonomic resolution to track pathogens at the species level, limiting clinical insights. Our study addresses this gap by validating ONT-based full-length (FL)-16S rRNA sequencing, a method that achieves species-level taxonomic precision critical for analyzing complex respiratory microbiomes. We benchmarked the microbiome composition of tracheal aspirates from ONT FL-16S rRNA workflows against Illumina V3-V4 data to demonstrate that long-read sequencing delivers comparable diversity profiles while resolving species-level diversity of clinically relevant species and microbes associated with the oral microbiome.
期刊介绍:
mSphere™ is a multi-disciplinary open-access journal that will focus on rapid publication of fundamental contributions to our understanding of microbiology. Its scope will reflect the immense range of fields within the microbial sciences, creating new opportunities for researchers to share findings that are transforming our understanding of human health and disease, ecosystems, neuroscience, agriculture, energy production, climate change, evolution, biogeochemical cycling, and food and drug production. Submissions will be encouraged of all high-quality work that makes fundamental contributions to our understanding of microbiology. mSphere™ will provide streamlined decisions, while carrying on ASM''s tradition for rigorous peer review.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


