{"title":"通过GPX4/NRF2途径,靶向prmt1介导的TAF15甲基化,通过抑制铁下沉来保护心肌梗死。","authors":"Guanshen Huang, Liwei He, Bishan Liang, Mingjian Gao, Jianming Huang, Hao Xia, Xinyu Li, Hai Li, Yunjun Ruan","doi":"10.1186/s13148-025-01935-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Acute myocardial infarction (AMI) remains a leading cause of morbidity and mortality worldwide. Ferroptosis, an iron-dependent form of regulated cell death, plays a crucial role in AMI progression. However, the molecular mechanisms regulating ferroptosis in AMI remain poorly understood. This study aims to investigate the role and potential regulatory mechanism of TAF15 in AMI.</p><p><strong>Methods: </strong>Bioinformatics analysis of gene expression datasets was conducted to identify differentially expressed genes in AMI samples. TAF15 expression was evaluated in clinical AMI patient blood samples, ischemia/reperfusion (I/R)-treated HL-1 cardiomyocytes, and myocardial tissues from the AMI mouse model using qRT-PCR and Western blot analyses. Gain- and loss-of-function experiments were performed to assess the effects of TAF15 and PRMT1 on myocardial injury, oxidative stress, and ferroptosis markers (Fe<sup>2</sup>⁺, MDA, GSH, GPX4, ROS) using electrocardiography, histopathology, CCK-8, EdU, TUNEL, ELISA, flow cytometry, qRT-PCR, and Western blot assays. Mechanistic studies, including luciferase reporter assays, chromatin immunoprecipitation (ChIP-qPCR), and bisulfite sequencing, were conducted to examine PRMT1-mediated TAF15 methylation and its regulatory effects.</p><p><strong>Results: </strong>TAF15 was significantly downregulated in AMI, as observed in patient samples and experimental models. Functionally, TAF15 overexpression significantly improved myocardial function by inhibiting ferroptosis. Notably, TAF15 overexpression restored GPX4 and NRF2 expression, reduced Fe<sup>2</sup>⁺ accumulation and lipid peroxidation (MDA levels), and increased GSH levels in both HL-1 cardiomyocytes and AMI mouse model. Mechanistic investigations revealed that TAF15 interacted with NRF2, enhancing TAF15 transcription and subsequently activating the GPX4/NRF2 axis, which protects against ferroptosis-induced cardiomyocyte death. Additionally, PRMT1 negatively regulated TAF15 via hypermethylation. PRMT1 knockdown significantly upregulated TAF15 expression, leading to reduced ferroptosis and improved cardiac function.</p><p><strong>Conclusions: </strong>This study establishes TAF15 as a novel regulator of ferroptosis in AMI, activating the GPX4/NRF2 pathway to mitigate oxidative stress and myocardial injury. Furthermore, PRMT1-mediated TAF15 hypermethylation promotes ferroptosis, thereby exacerbating myocardial damage. These findings suggest that targeting the PRMT1/TAF15/GPX4-NRF2 axis represents a promising therapeutic strategy for AMI treatment by inhibiting ferroptotic cell death and improving cardiac function.</p>","PeriodicalId":10366,"journal":{"name":"Clinical Epigenetics","volume":"17 1","pages":"129"},"PeriodicalIF":4.4000,"publicationDate":"2025-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12285139/pdf/","citationCount":"0","resultStr":"{\"title\":\"Targeting PRMT1-mediated methylation of TAF15 to protect against myocardial infarction by inhibiting ferroptosis via the GPX4/NRF2 pathway.\",\"authors\":\"Guanshen Huang, Liwei He, Bishan Liang, Mingjian Gao, Jianming Huang, Hao Xia, Xinyu Li, Hai Li, Yunjun Ruan\",\"doi\":\"10.1186/s13148-025-01935-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Acute myocardial infarction (AMI) remains a leading cause of morbidity and mortality worldwide. Ferroptosis, an iron-dependent form of regulated cell death, plays a crucial role in AMI progression. However, the molecular mechanisms regulating ferroptosis in AMI remain poorly understood. This study aims to investigate the role and potential regulatory mechanism of TAF15 in AMI.</p><p><strong>Methods: </strong>Bioinformatics analysis of gene expression datasets was conducted to identify differentially expressed genes in AMI samples. TAF15 expression was evaluated in clinical AMI patient blood samples, ischemia/reperfusion (I/R)-treated HL-1 cardiomyocytes, and myocardial tissues from the AMI mouse model using qRT-PCR and Western blot analyses. Gain- and loss-of-function experiments were performed to assess the effects of TAF15 and PRMT1 on myocardial injury, oxidative stress, and ferroptosis markers (Fe<sup>2</sup>⁺, MDA, GSH, GPX4, ROS) using electrocardiography, histopathology, CCK-8, EdU, TUNEL, ELISA, flow cytometry, qRT-PCR, and Western blot assays. Mechanistic studies, including luciferase reporter assays, chromatin immunoprecipitation (ChIP-qPCR), and bisulfite sequencing, were conducted to examine PRMT1-mediated TAF15 methylation and its regulatory effects.</p><p><strong>Results: </strong>TAF15 was significantly downregulated in AMI, as observed in patient samples and experimental models. Functionally, TAF15 overexpression significantly improved myocardial function by inhibiting ferroptosis. Notably, TAF15 overexpression restored GPX4 and NRF2 expression, reduced Fe<sup>2</sup>⁺ accumulation and lipid peroxidation (MDA levels), and increased GSH levels in both HL-1 cardiomyocytes and AMI mouse model. Mechanistic investigations revealed that TAF15 interacted with NRF2, enhancing TAF15 transcription and subsequently activating the GPX4/NRF2 axis, which protects against ferroptosis-induced cardiomyocyte death. Additionally, PRMT1 negatively regulated TAF15 via hypermethylation. PRMT1 knockdown significantly upregulated TAF15 expression, leading to reduced ferroptosis and improved cardiac function.</p><p><strong>Conclusions: </strong>This study establishes TAF15 as a novel regulator of ferroptosis in AMI, activating the GPX4/NRF2 pathway to mitigate oxidative stress and myocardial injury. Furthermore, PRMT1-mediated TAF15 hypermethylation promotes ferroptosis, thereby exacerbating myocardial damage. These findings suggest that targeting the PRMT1/TAF15/GPX4-NRF2 axis represents a promising therapeutic strategy for AMI treatment by inhibiting ferroptotic cell death and improving cardiac function.</p>\",\"PeriodicalId\":10366,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"17 1\",\"pages\":\"129\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-07-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12285139/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-025-01935-8\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-025-01935-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Targeting PRMT1-mediated methylation of TAF15 to protect against myocardial infarction by inhibiting ferroptosis via the GPX4/NRF2 pathway.
Background: Acute myocardial infarction (AMI) remains a leading cause of morbidity and mortality worldwide. Ferroptosis, an iron-dependent form of regulated cell death, plays a crucial role in AMI progression. However, the molecular mechanisms regulating ferroptosis in AMI remain poorly understood. This study aims to investigate the role and potential regulatory mechanism of TAF15 in AMI.
Methods: Bioinformatics analysis of gene expression datasets was conducted to identify differentially expressed genes in AMI samples. TAF15 expression was evaluated in clinical AMI patient blood samples, ischemia/reperfusion (I/R)-treated HL-1 cardiomyocytes, and myocardial tissues from the AMI mouse model using qRT-PCR and Western blot analyses. Gain- and loss-of-function experiments were performed to assess the effects of TAF15 and PRMT1 on myocardial injury, oxidative stress, and ferroptosis markers (Fe2⁺, MDA, GSH, GPX4, ROS) using electrocardiography, histopathology, CCK-8, EdU, TUNEL, ELISA, flow cytometry, qRT-PCR, and Western blot assays. Mechanistic studies, including luciferase reporter assays, chromatin immunoprecipitation (ChIP-qPCR), and bisulfite sequencing, were conducted to examine PRMT1-mediated TAF15 methylation and its regulatory effects.
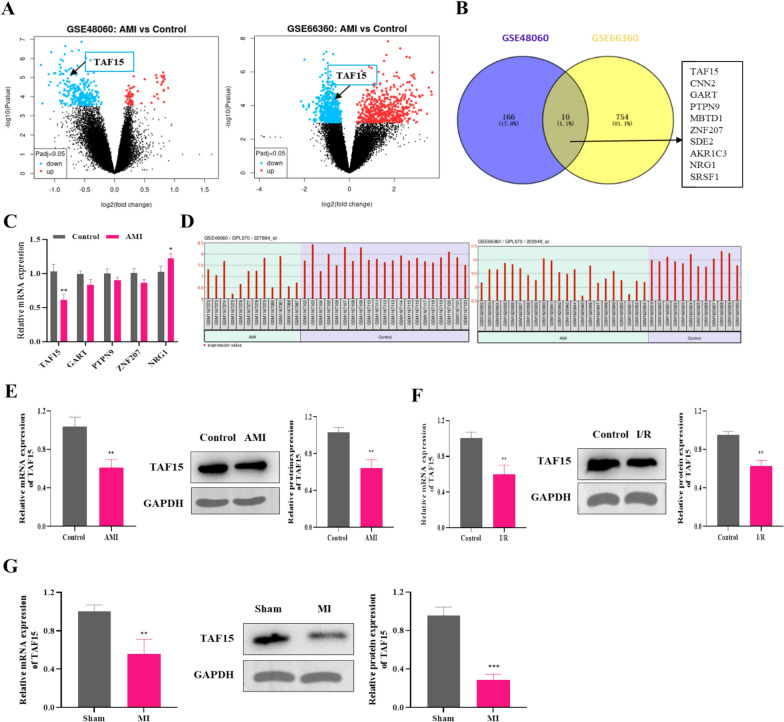
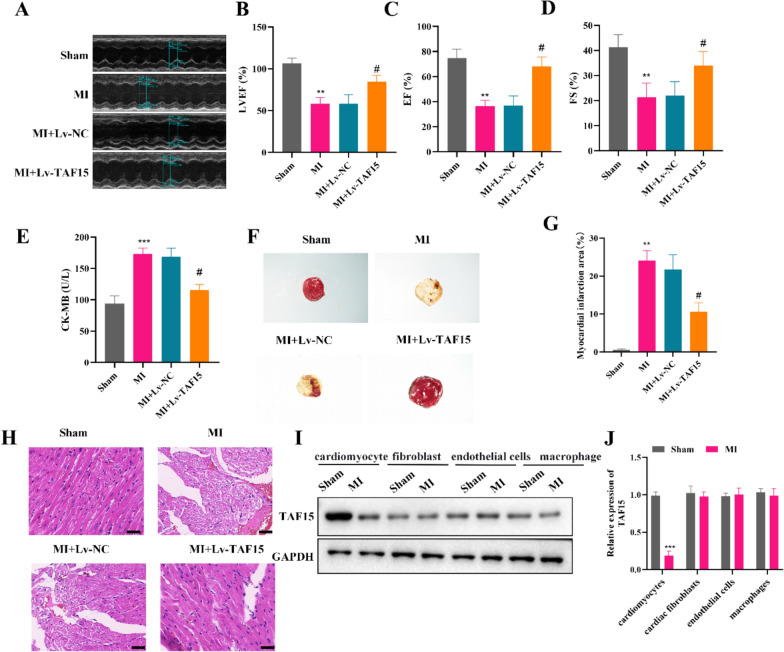
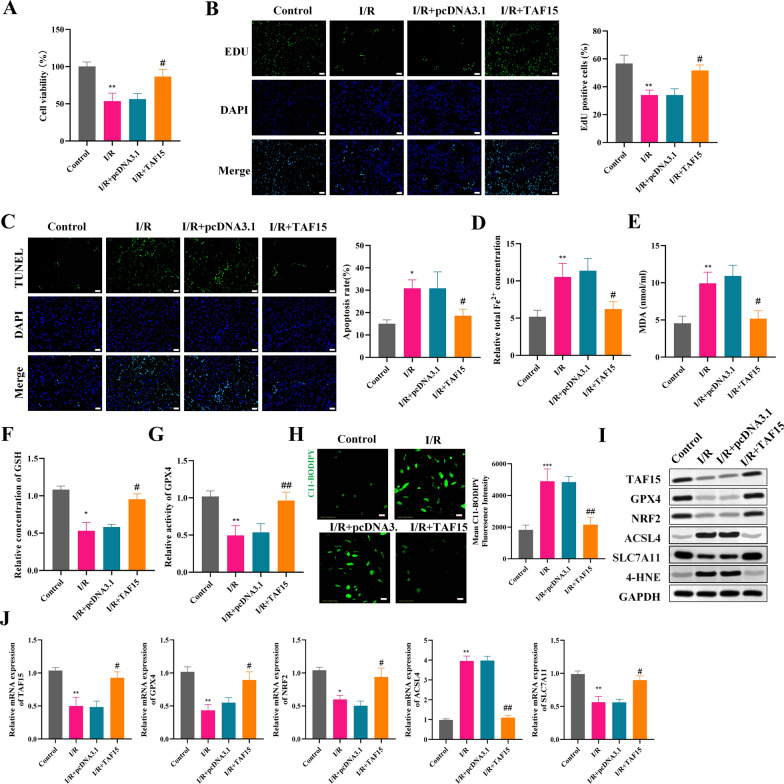
Results: TAF15 was significantly downregulated in AMI, as observed in patient samples and experimental models. Functionally, TAF15 overexpression significantly improved myocardial function by inhibiting ferroptosis. Notably, TAF15 overexpression restored GPX4 and NRF2 expression, reduced Fe2⁺ accumulation and lipid peroxidation (MDA levels), and increased GSH levels in both HL-1 cardiomyocytes and AMI mouse model. Mechanistic investigations revealed that TAF15 interacted with NRF2, enhancing TAF15 transcription and subsequently activating the GPX4/NRF2 axis, which protects against ferroptosis-induced cardiomyocyte death. Additionally, PRMT1 negatively regulated TAF15 via hypermethylation. PRMT1 knockdown significantly upregulated TAF15 expression, leading to reduced ferroptosis and improved cardiac function.
Conclusions: This study establishes TAF15 as a novel regulator of ferroptosis in AMI, activating the GPX4/NRF2 pathway to mitigate oxidative stress and myocardial injury. Furthermore, PRMT1-mediated TAF15 hypermethylation promotes ferroptosis, thereby exacerbating myocardial damage. These findings suggest that targeting the PRMT1/TAF15/GPX4-NRF2 axis represents a promising therapeutic strategy for AMI treatment by inhibiting ferroptotic cell death and improving cardiac function.
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


