{"title":"Aryana-bs:亚硫酸酯测序读取的上下文感知校准。","authors":"Hassan Nikaein, Ali Sharifi-Zarchi, Afsoon Afzal, Saeedeh Ezzati, Farzane Rasti, Hamidreza Chitsaz, Govindarajan Kunde-Ramamoorthy","doi":"10.1186/s12859-025-06182-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>DNA methylation is essential in various biological processes, including imprinting, development, inflammation, and numerous disorders, such as cancer. Bisulfite sequencing (BS) serves as the gold standard for measuring DNA methylation at single-base resolution by converting unmethylated cytosines to thymines while leaving methylated cytosines intact. However, this C-to-T conversion presents a well-known challenge in conventional short-read aligners, which treat these conversions as substitutions. Many aligners that require seed sequences fail when frequent C-to-T conversions occur over short distances, resulting in reduced alignment accuracy. To address this challenge, two alignment methods have been well established: three-letter alignment and wildcard alignment. Three-letter alignment faces the significant issue of data loss by converting all thymines to cytosines, which obscures meaningful information. On the other hand, wildcard alignment introduces a biased alignment, failing to treat reads from unmethylated and methylated regions equally, leading to artifacts in methylation level estimation and inaccuracies in quantifying DNA methylation. This work introduces ARYANA-BS, a novel BS aligner that diverges from conventional DNA aligners by directly integrating BS-specific base alterations within its alignment engine. Leveraging known DNA methylation patterns across different genomic contexts, ARYANA-BS constructs five indexes from the reference genome, aligns each read to all indexes, and selects the alignment with the minimum penalty. To further refine alignment accuracy, an optional Expectation-Maximization (EM) step is incorporated, which integrates methylation probability information into the decision-making process for choosing the optimal index for each read. This approach aims to enhance BS read alignment accuracy by accommodating the complexities of DNA methylation patterns across diverse genomic contexts.</p><p><strong>Results: </strong>Experimental evaluations on both simulated and real data reveal that ARYANA-BS achieves state-of-the-art accuracy, maintaining competitive speed and memory efficiency.</p><p><strong>Conclusions: </strong>ARYANA-BS significantly improves alignment accuracy for bisulfite sequencing data by effectively integrating DNA methylation-specific alterations and genomic context. It outperforms existing methods, such as BSMAP, bwa-meth, Bismark, BSBolt, and abismal, particularly in robustness against genomic biases and alignment of longer, higher-error reads, demonstrating suitability for cancer research and cell-free DNA studies. While the Expectation-Maximization (EM) algorithm provides only modest initial improvements, it establishes a valuable framework for future refinement and potential enhancements in sensitive applications.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"188"},"PeriodicalIF":3.3000,"publicationDate":"2025-07-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12281798/pdf/","citationCount":"0","resultStr":"{\"title\":\"Aryana-bs: context-aware alignment of bisulfite-sequencing reads.\",\"authors\":\"Hassan Nikaein, Ali Sharifi-Zarchi, Afsoon Afzal, Saeedeh Ezzati, Farzane Rasti, Hamidreza Chitsaz, Govindarajan Kunde-Ramamoorthy\",\"doi\":\"10.1186/s12859-025-06182-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>DNA methylation is essential in various biological processes, including imprinting, development, inflammation, and numerous disorders, such as cancer. Bisulfite sequencing (BS) serves as the gold standard for measuring DNA methylation at single-base resolution by converting unmethylated cytosines to thymines while leaving methylated cytosines intact. However, this C-to-T conversion presents a well-known challenge in conventional short-read aligners, which treat these conversions as substitutions. Many aligners that require seed sequences fail when frequent C-to-T conversions occur over short distances, resulting in reduced alignment accuracy. To address this challenge, two alignment methods have been well established: three-letter alignment and wildcard alignment. Three-letter alignment faces the significant issue of data loss by converting all thymines to cytosines, which obscures meaningful information. On the other hand, wildcard alignment introduces a biased alignment, failing to treat reads from unmethylated and methylated regions equally, leading to artifacts in methylation level estimation and inaccuracies in quantifying DNA methylation. This work introduces ARYANA-BS, a novel BS aligner that diverges from conventional DNA aligners by directly integrating BS-specific base alterations within its alignment engine. Leveraging known DNA methylation patterns across different genomic contexts, ARYANA-BS constructs five indexes from the reference genome, aligns each read to all indexes, and selects the alignment with the minimum penalty. To further refine alignment accuracy, an optional Expectation-Maximization (EM) step is incorporated, which integrates methylation probability information into the decision-making process for choosing the optimal index for each read. This approach aims to enhance BS read alignment accuracy by accommodating the complexities of DNA methylation patterns across diverse genomic contexts.</p><p><strong>Results: </strong>Experimental evaluations on both simulated and real data reveal that ARYANA-BS achieves state-of-the-art accuracy, maintaining competitive speed and memory efficiency.</p><p><strong>Conclusions: </strong>ARYANA-BS significantly improves alignment accuracy for bisulfite sequencing data by effectively integrating DNA methylation-specific alterations and genomic context. It outperforms existing methods, such as BSMAP, bwa-meth, Bismark, BSBolt, and abismal, particularly in robustness against genomic biases and alignment of longer, higher-error reads, demonstrating suitability for cancer research and cell-free DNA studies. While the Expectation-Maximization (EM) algorithm provides only modest initial improvements, it establishes a valuable framework for future refinement and potential enhancements in sensitive applications.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"188\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-07-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12281798/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06182-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06182-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Aryana-bs: context-aware alignment of bisulfite-sequencing reads.
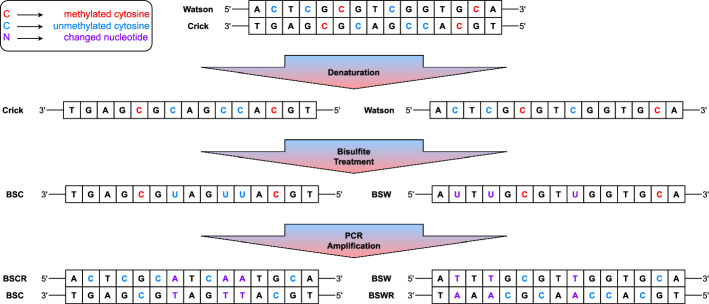
Background: DNA methylation is essential in various biological processes, including imprinting, development, inflammation, and numerous disorders, such as cancer. Bisulfite sequencing (BS) serves as the gold standard for measuring DNA methylation at single-base resolution by converting unmethylated cytosines to thymines while leaving methylated cytosines intact. However, this C-to-T conversion presents a well-known challenge in conventional short-read aligners, which treat these conversions as substitutions. Many aligners that require seed sequences fail when frequent C-to-T conversions occur over short distances, resulting in reduced alignment accuracy. To address this challenge, two alignment methods have been well established: three-letter alignment and wildcard alignment. Three-letter alignment faces the significant issue of data loss by converting all thymines to cytosines, which obscures meaningful information. On the other hand, wildcard alignment introduces a biased alignment, failing to treat reads from unmethylated and methylated regions equally, leading to artifacts in methylation level estimation and inaccuracies in quantifying DNA methylation. This work introduces ARYANA-BS, a novel BS aligner that diverges from conventional DNA aligners by directly integrating BS-specific base alterations within its alignment engine. Leveraging known DNA methylation patterns across different genomic contexts, ARYANA-BS constructs five indexes from the reference genome, aligns each read to all indexes, and selects the alignment with the minimum penalty. To further refine alignment accuracy, an optional Expectation-Maximization (EM) step is incorporated, which integrates methylation probability information into the decision-making process for choosing the optimal index for each read. This approach aims to enhance BS read alignment accuracy by accommodating the complexities of DNA methylation patterns across diverse genomic contexts.
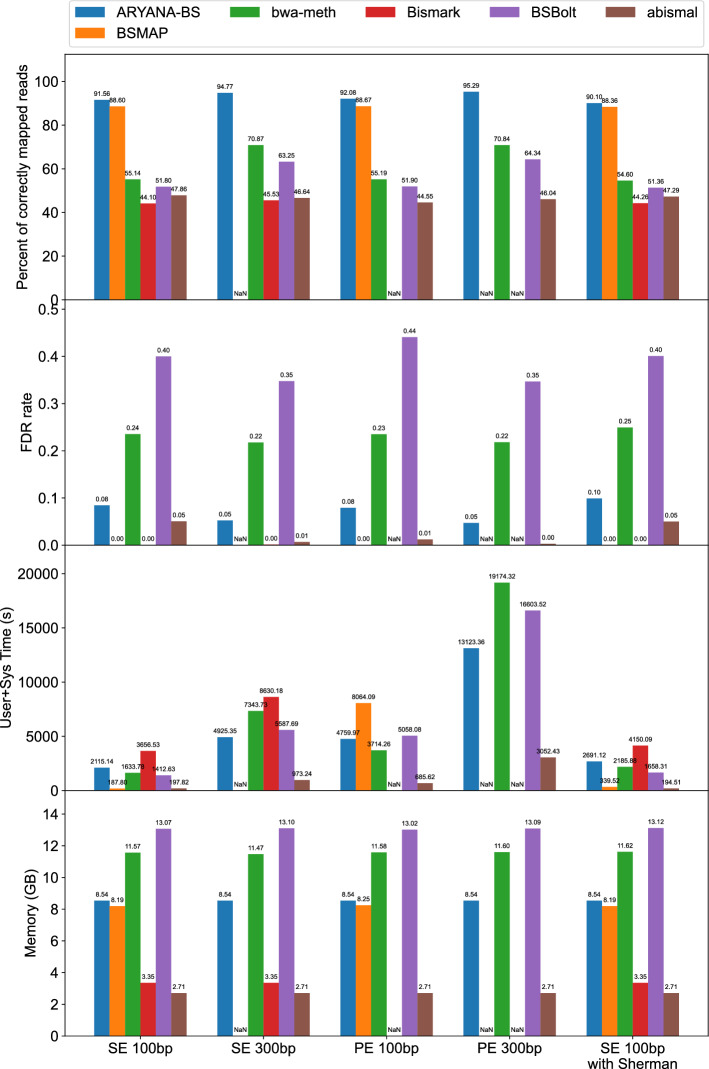
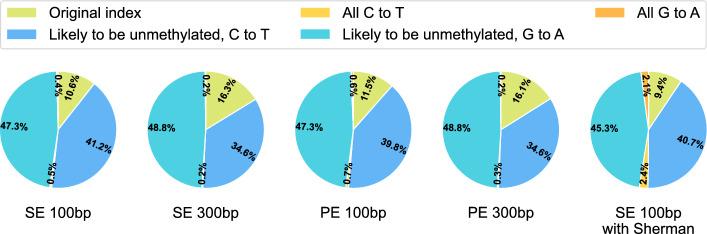
Results: Experimental evaluations on both simulated and real data reveal that ARYANA-BS achieves state-of-the-art accuracy, maintaining competitive speed and memory efficiency.
Conclusions: ARYANA-BS significantly improves alignment accuracy for bisulfite sequencing data by effectively integrating DNA methylation-specific alterations and genomic context. It outperforms existing methods, such as BSMAP, bwa-meth, Bismark, BSBolt, and abismal, particularly in robustness against genomic biases and alignment of longer, higher-error reads, demonstrating suitability for cancer research and cell-free DNA studies. While the Expectation-Maximization (EM) algorithm provides only modest initial improvements, it establishes a valuable framework for future refinement and potential enhancements in sensitive applications.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


