{"title":"MitSorter:基于差异甲基化的mtDNA和NuMT - ONT读取准确区分的独立工具。","authors":"Sharon Natasha Cox, Angelo Sante Varvara, Graziano Pesole","doi":"10.1093/bioadv/vbaf135","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The accurate differentiation between mitochondrial DNA (mtDNA) and nuclear mitochondrial DNA segments (NuMTs) is a critical challenge in studies involving mitochondrial disorders. Mapping the mtDNA mutation spectrum and quantifying heteroplasmy are complex tasks when using next-generation sequencing methods, mostly due to NuMTs contamination in data analysis.</p><p><strong>Results: </strong>Here, we present a novel, easy-to-use standalone command-line tool designed to reliably discriminate long reads originated by either mtDNA or NuMTs and generated by Oxford Nanopore Technologies (ONT) sequencing based on the known lack of CpG methylation in human mtDNA. MitSorter aligns the reads to the mitochondrial genome incorporating base modification calls directly from raw POD5 files. The resulting BAM file is then partitioned into two separate BAM files: one containing unmethylated reads and the other containing methylated reads. We show that MitSorter analysis can provide a more accurate landscape of the mtDNA mutation profile. We describe here the tool's features, computational framework, validation approach, and its potential applications in other genomic research areas.</p><p><strong>Availability and implementation: </strong>Source code and documentation, are available at https://github.com/asvarvara/MitSorter.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf135"},"PeriodicalIF":2.8000,"publicationDate":"2025-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12275464/pdf/","citationCount":"0","resultStr":"{\"title\":\"MitSorter: a standalone tool for accurate discrimination of mtDNA and NuMT ONT reads based on differential methylation.\",\"authors\":\"Sharon Natasha Cox, Angelo Sante Varvara, Graziano Pesole\",\"doi\":\"10.1093/bioadv/vbaf135\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>The accurate differentiation between mitochondrial DNA (mtDNA) and nuclear mitochondrial DNA segments (NuMTs) is a critical challenge in studies involving mitochondrial disorders. Mapping the mtDNA mutation spectrum and quantifying heteroplasmy are complex tasks when using next-generation sequencing methods, mostly due to NuMTs contamination in data analysis.</p><p><strong>Results: </strong>Here, we present a novel, easy-to-use standalone command-line tool designed to reliably discriminate long reads originated by either mtDNA or NuMTs and generated by Oxford Nanopore Technologies (ONT) sequencing based on the known lack of CpG methylation in human mtDNA. MitSorter aligns the reads to the mitochondrial genome incorporating base modification calls directly from raw POD5 files. The resulting BAM file is then partitioned into two separate BAM files: one containing unmethylated reads and the other containing methylated reads. We show that MitSorter analysis can provide a more accurate landscape of the mtDNA mutation profile. We describe here the tool's features, computational framework, validation approach, and its potential applications in other genomic research areas.</p><p><strong>Availability and implementation: </strong>Source code and documentation, are available at https://github.com/asvarvara/MitSorter.</p>\",\"PeriodicalId\":72368,\"journal\":{\"name\":\"Bioinformatics advances\",\"volume\":\"5 1\",\"pages\":\"vbaf135\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12275464/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioadv/vbaf135\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf135","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
MitSorter: a standalone tool for accurate discrimination of mtDNA and NuMT ONT reads based on differential methylation.
Motivation: The accurate differentiation between mitochondrial DNA (mtDNA) and nuclear mitochondrial DNA segments (NuMTs) is a critical challenge in studies involving mitochondrial disorders. Mapping the mtDNA mutation spectrum and quantifying heteroplasmy are complex tasks when using next-generation sequencing methods, mostly due to NuMTs contamination in data analysis.
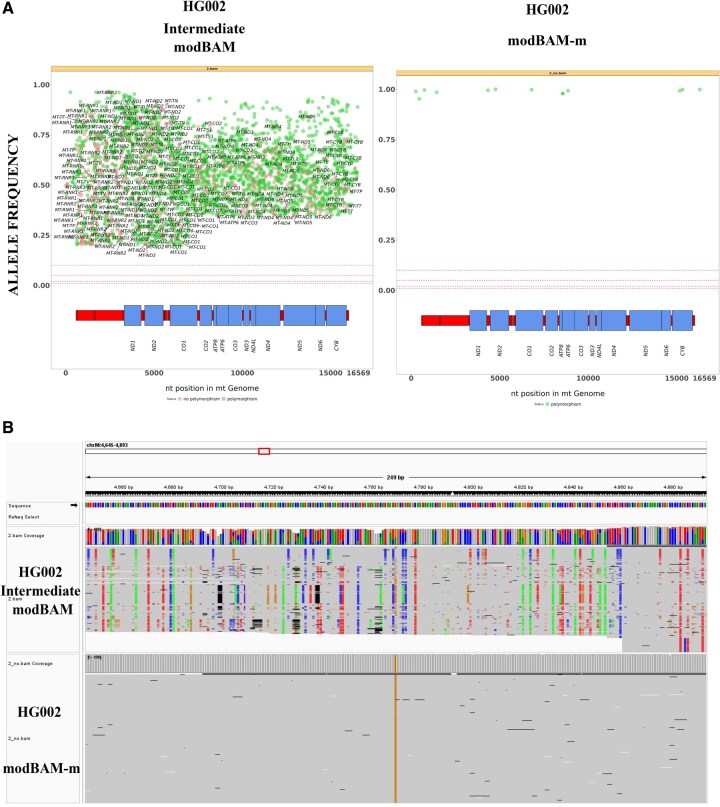
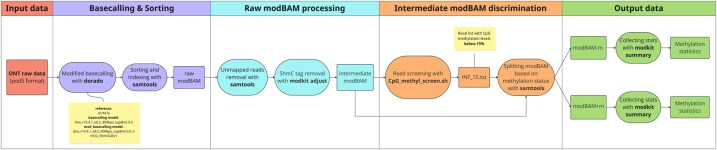
Results: Here, we present a novel, easy-to-use standalone command-line tool designed to reliably discriminate long reads originated by either mtDNA or NuMTs and generated by Oxford Nanopore Technologies (ONT) sequencing based on the known lack of CpG methylation in human mtDNA. MitSorter aligns the reads to the mitochondrial genome incorporating base modification calls directly from raw POD5 files. The resulting BAM file is then partitioned into two separate BAM files: one containing unmethylated reads and the other containing methylated reads. We show that MitSorter analysis can provide a more accurate landscape of the mtDNA mutation profile. We describe here the tool's features, computational framework, validation approach, and its potential applications in other genomic research areas.
Availability and implementation: Source code and documentation, are available at https://github.com/asvarvara/MitSorter.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


