{"title":"由GRIN2A基因移码突变引起的神经发育障碍:一例报告。","authors":"Chen Xu, Man-Li Wang, Wei-Hao Ling, Ji-Hong Tang","doi":"10.21037/tp-2025-93","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pathogenic variants in <i>GRIN2A</i>, encoding the GluN2A subunit of the N-methyl-D-aspartate receptor (NMDAR), are increasingly recognized as causes of neurodevelopmental disorders, particularly within the epilepsy-aphasia spectrum. However, presentations without clinical seizures-especially those initially manifesting as isolated ataxia-are rarely reported. We describe a previously unreported <i>GRIN2A</i> frameshift variant associated with early-onset ataxia, delayed-onset electrographic abnormalities, and favorable response to immunotherapy.</p><p><strong>Case description: </strong>A 23-month-old boy presented with subacute gait ataxia following a viral illness. Neuroimaging, cerebrospinal fluid analysis, and an extensive autoimmune panel were unremarkable. Initial immunotherapy with high-dose corticosteroids and intravenous immunoglobulin (IVIG) led to transient improvement. Five months later, he developed recurrent ataxia, speech regression, drooling, and global developmental delay, still without overt seizures. Video electroencephalogram (EEG) revealed electrical status epilepticus during slow-wave sleep (ESES) with a spike-wave index exceeding 85%. Trio-based whole genome sequencing identified a novel heterozygous frameshift variant in <i>GRIN2A</i> (c.1717delG, p.Val573Phefs*16), predicted to result in loss of all transmembrane domains. Repeat immunotherapy produced significant clinical improvement, including restored ambulation, cessation of drooling, enhanced speech output, and marked reduction in epileptiform discharges. The patient remained seizure-free during the reported treatment period. Notably, his mother, a carrier of the same variant, reported only a brief history of childhood seizures with minimal residual speech disturbance.</p><p><strong>Conclusions: </strong>This case expands the phenotypic spectrum of <i>GRIN2A</i>-related disorders to include early isolated ataxia and delayed electrographic epilepsy in the absence of clinical seizures. It highlights the diagnostic value of early genetic testing in atypical neurodevelopmental syndromes and suggests that immunotherapy may confer clinical and electrophysiological benefits, even in presumed NMDAR loss-of-function states. Integration of genomics, neurophysiology, and immune-modulating strategies may inform future precision therapies for <i>GRIN2A</i>-associated encephalopathies.</p>","PeriodicalId":23294,"journal":{"name":"Translational pediatrics","volume":"14 6","pages":"1353-1361"},"PeriodicalIF":1.7000,"publicationDate":"2025-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12268631/pdf/","citationCount":"0","resultStr":"{\"title\":\"Neurodevelopmental disorder due to a frameshift mutation in the <i>GRIN2A</i> gene: a case report.\",\"authors\":\"Chen Xu, Man-Li Wang, Wei-Hao Ling, Ji-Hong Tang\",\"doi\":\"10.21037/tp-2025-93\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Pathogenic variants in <i>GRIN2A</i>, encoding the GluN2A subunit of the N-methyl-D-aspartate receptor (NMDAR), are increasingly recognized as causes of neurodevelopmental disorders, particularly within the epilepsy-aphasia spectrum. However, presentations without clinical seizures-especially those initially manifesting as isolated ataxia-are rarely reported. We describe a previously unreported <i>GRIN2A</i> frameshift variant associated with early-onset ataxia, delayed-onset electrographic abnormalities, and favorable response to immunotherapy.</p><p><strong>Case description: </strong>A 23-month-old boy presented with subacute gait ataxia following a viral illness. Neuroimaging, cerebrospinal fluid analysis, and an extensive autoimmune panel were unremarkable. Initial immunotherapy with high-dose corticosteroids and intravenous immunoglobulin (IVIG) led to transient improvement. Five months later, he developed recurrent ataxia, speech regression, drooling, and global developmental delay, still without overt seizures. Video electroencephalogram (EEG) revealed electrical status epilepticus during slow-wave sleep (ESES) with a spike-wave index exceeding 85%. Trio-based whole genome sequencing identified a novel heterozygous frameshift variant in <i>GRIN2A</i> (c.1717delG, p.Val573Phefs*16), predicted to result in loss of all transmembrane domains. Repeat immunotherapy produced significant clinical improvement, including restored ambulation, cessation of drooling, enhanced speech output, and marked reduction in epileptiform discharges. The patient remained seizure-free during the reported treatment period. Notably, his mother, a carrier of the same variant, reported only a brief history of childhood seizures with minimal residual speech disturbance.</p><p><strong>Conclusions: </strong>This case expands the phenotypic spectrum of <i>GRIN2A</i>-related disorders to include early isolated ataxia and delayed electrographic epilepsy in the absence of clinical seizures. It highlights the diagnostic value of early genetic testing in atypical neurodevelopmental syndromes and suggests that immunotherapy may confer clinical and electrophysiological benefits, even in presumed NMDAR loss-of-function states. Integration of genomics, neurophysiology, and immune-modulating strategies may inform future precision therapies for <i>GRIN2A</i>-associated encephalopathies.</p>\",\"PeriodicalId\":23294,\"journal\":{\"name\":\"Translational pediatrics\",\"volume\":\"14 6\",\"pages\":\"1353-1361\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12268631/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational pediatrics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tp-2025-93\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/6/18 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tp-2025-93","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/18 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
摘要
背景:编码n -甲基- d -天冬氨酸受体(NMDAR) GluN2A亚基的GRIN2A致病性变异越来越多地被认为是神经发育障碍的原因,特别是在癫痫-失语症谱系中。然而,没有临床发作的表现-特别是那些最初表现为孤立性共济失调的-很少被报道。我们描述了一种以前未报道的GRIN2A移码变异与早发性共济失调、迟发性电异常和对免疫治疗的有利反应相关。病例描述:一个23个月大的男孩在病毒性疾病后出现亚急性步态共济失调。神经影像学、脑脊液分析和广泛的自身免疫检查都没有异常。最初使用高剂量皮质类固醇和静脉注射免疫球蛋白(IVIG)的免疫治疗导致短暂的改善。5个月后,患者出现复发性共济失调、语言减退、流口水和整体发育迟缓,但仍无明显癫痫发作。视频脑电图(EEG)显示慢波睡眠(ESES)期间的电性癫痫持续状态,其峰波指数超过85%。三基全基因组测序在GRIN2A中发现了一个新的杂合移码变异(c.1717delG, p.Val573Phefs*16),预计会导致所有跨膜结构域的丢失。重复免疫治疗产生了显著的临床改善,包括恢复行走、停止流口水、增强语言输出和癫痫样放电的显著减少。在报告的治疗期间,患者保持无癫痫发作。值得注意的是,他的母亲是同一变异的携带者,据报道,她只有短暂的童年癫痫发作史,并伴有轻微的残余语言障碍。结论:该病例扩大了grin2a相关疾病的表型谱,包括早期孤立性共济失调和没有临床发作的延迟性电性癫痫。它强调了早期基因检测在非典型神经发育综合征中的诊断价值,并表明免疫治疗可能会带来临床和电生理上的益处,即使在假定的NMDAR功能丧失状态下也是如此。基因组学、神经生理学和免疫调节策略的整合可能为未来针对grin2a相关脑病的精确治疗提供信息。
Neurodevelopmental disorder due to a frameshift mutation in the GRIN2A gene: a case report.
Background: Pathogenic variants in GRIN2A, encoding the GluN2A subunit of the N-methyl-D-aspartate receptor (NMDAR), are increasingly recognized as causes of neurodevelopmental disorders, particularly within the epilepsy-aphasia spectrum. However, presentations without clinical seizures-especially those initially manifesting as isolated ataxia-are rarely reported. We describe a previously unreported GRIN2A frameshift variant associated with early-onset ataxia, delayed-onset electrographic abnormalities, and favorable response to immunotherapy.
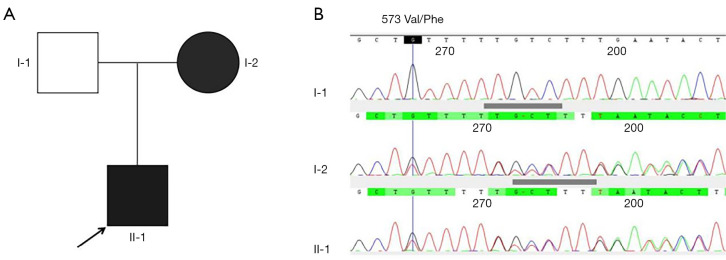
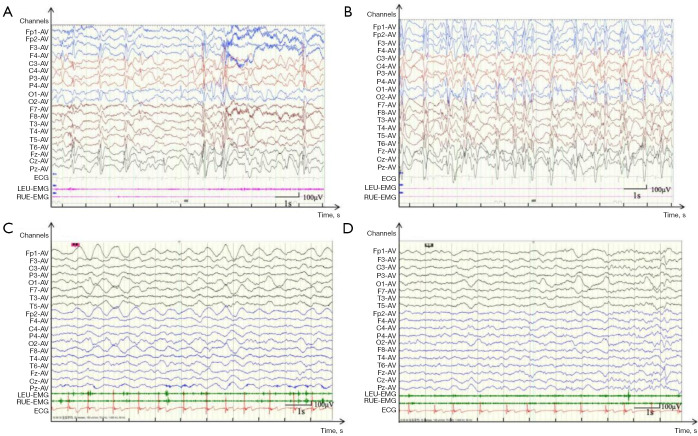
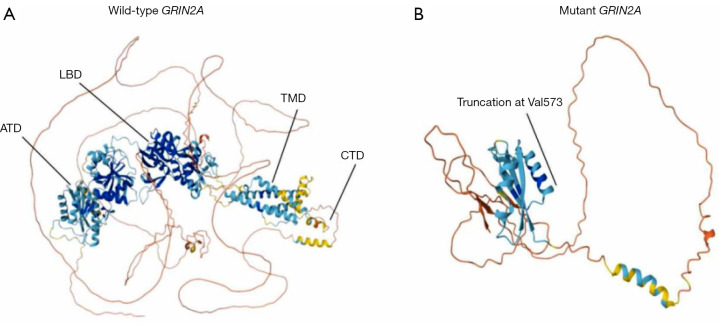
Case description: A 23-month-old boy presented with subacute gait ataxia following a viral illness. Neuroimaging, cerebrospinal fluid analysis, and an extensive autoimmune panel were unremarkable. Initial immunotherapy with high-dose corticosteroids and intravenous immunoglobulin (IVIG) led to transient improvement. Five months later, he developed recurrent ataxia, speech regression, drooling, and global developmental delay, still without overt seizures. Video electroencephalogram (EEG) revealed electrical status epilepticus during slow-wave sleep (ESES) with a spike-wave index exceeding 85%. Trio-based whole genome sequencing identified a novel heterozygous frameshift variant in GRIN2A (c.1717delG, p.Val573Phefs*16), predicted to result in loss of all transmembrane domains. Repeat immunotherapy produced significant clinical improvement, including restored ambulation, cessation of drooling, enhanced speech output, and marked reduction in epileptiform discharges. The patient remained seizure-free during the reported treatment period. Notably, his mother, a carrier of the same variant, reported only a brief history of childhood seizures with minimal residual speech disturbance.
Conclusions: This case expands the phenotypic spectrum of GRIN2A-related disorders to include early isolated ataxia and delayed electrographic epilepsy in the absence of clinical seizures. It highlights the diagnostic value of early genetic testing in atypical neurodevelopmental syndromes and suggests that immunotherapy may confer clinical and electrophysiological benefits, even in presumed NMDAR loss-of-function states. Integration of genomics, neurophysiology, and immune-modulating strategies may inform future precision therapies for GRIN2A-associated encephalopathies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


