Ilya S Dantsev, Anastasiia A Buianova, Ekaterina B Polykova, Ekaterina A Nikolaeva, Evgenii V Vasilyev, Angelina Iu Yakshina, Mariia A Parfenenko, Mariia I Yablonskaya, Oksana S Kurinnaia, Ivan Yu Iourov
{"title":"Xp22.31缺失导致的心律失常、发育迟缓、癫痫、鱼鳞病:文献复习及病例报告","authors":"Ilya S Dantsev, Anastasiia A Buianova, Ekaterina B Polykova, Ekaterina A Nikolaeva, Evgenii V Vasilyev, Angelina Iu Yakshina, Mariia A Parfenenko, Mariia I Yablonskaya, Oksana S Kurinnaia, Ivan Yu Iourov","doi":"10.21037/tp-2025-87","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The broad phenotypic variability observed in patients with Xp22.31 deletion, traditionally associated with X-linked ichthyosis (XLI), is increasingly recognized as encompassing a wider spectrum of clinical manifestations. While ichthyosis, caused by <i>STS</i> gene deletion, remains the hallmark feature, recent studies and genomic analyses (e.g., chromosomal microarray and whole genome sequencing) have revealed additional extracutaneous phenotypes. These include corneal opacification, cryptorchidism, autism spectrum disorders, intellectual disability, epilepsy, developmental delay, renal anomalies, and an elevated risk of atrial fibrillation and other cardiac arrhythmias, particularly in males. Interestingly, duplications of this region are usually considered benign, underscoring the need for nuanced interpretation.</p><p><strong>Case description: </strong>We describe three unrelated male patients carrying hemizygous Xp22.31 microdeletions (~1.6 Mb), all presenting with mild to moderate ichthyosis characterized by \"plate-like\" desquamation. Two patients exhibited intellectual disability and bradyarrhythmia, while one experienced seizures. None had major congenital anomalies, and all underwent chromosomal microarray analysis to confirm the diagnosis.</p><p><strong>Conclusions: </strong>Our findings emphasize the need for a multidisciplinary approach when evaluating patients with Xp22.31 deletions, extending beyond dermatologic assessment to include neurological and cardiological evaluations, even in the absence of overt symptoms. This broader phenotypic understanding may enhance clinical management, support more accurate genetic counseling, and inform prenatal diagnostic decision-making. Furthermore, our observations support the hypothesis that genes within the deleted region-such as <i>STS</i>, <i>PNPLA4</i>, and <i>VCX</i> family genes-may contribute to the pathogenesis of neurological and cardiac abnormalities, warranting further functional studies and long-term clinical monitoring.</p>","PeriodicalId":23294,"journal":{"name":"Translational pediatrics","volume":"14 6","pages":"1370-1379"},"PeriodicalIF":1.7000,"publicationDate":"2025-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12268512/pdf/","citationCount":"0","resultStr":"{\"title\":\"Cardiac arrhythmia, developmental delay, epilepsy and ichthyosis due to Xp22.31 deletion: review of literature and case report.\",\"authors\":\"Ilya S Dantsev, Anastasiia A Buianova, Ekaterina B Polykova, Ekaterina A Nikolaeva, Evgenii V Vasilyev, Angelina Iu Yakshina, Mariia A Parfenenko, Mariia I Yablonskaya, Oksana S Kurinnaia, Ivan Yu Iourov\",\"doi\":\"10.21037/tp-2025-87\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The broad phenotypic variability observed in patients with Xp22.31 deletion, traditionally associated with X-linked ichthyosis (XLI), is increasingly recognized as encompassing a wider spectrum of clinical manifestations. While ichthyosis, caused by <i>STS</i> gene deletion, remains the hallmark feature, recent studies and genomic analyses (e.g., chromosomal microarray and whole genome sequencing) have revealed additional extracutaneous phenotypes. These include corneal opacification, cryptorchidism, autism spectrum disorders, intellectual disability, epilepsy, developmental delay, renal anomalies, and an elevated risk of atrial fibrillation and other cardiac arrhythmias, particularly in males. Interestingly, duplications of this region are usually considered benign, underscoring the need for nuanced interpretation.</p><p><strong>Case description: </strong>We describe three unrelated male patients carrying hemizygous Xp22.31 microdeletions (~1.6 Mb), all presenting with mild to moderate ichthyosis characterized by \\\"plate-like\\\" desquamation. Two patients exhibited intellectual disability and bradyarrhythmia, while one experienced seizures. None had major congenital anomalies, and all underwent chromosomal microarray analysis to confirm the diagnosis.</p><p><strong>Conclusions: </strong>Our findings emphasize the need for a multidisciplinary approach when evaluating patients with Xp22.31 deletions, extending beyond dermatologic assessment to include neurological and cardiological evaluations, even in the absence of overt symptoms. This broader phenotypic understanding may enhance clinical management, support more accurate genetic counseling, and inform prenatal diagnostic decision-making. Furthermore, our observations support the hypothesis that genes within the deleted region-such as <i>STS</i>, <i>PNPLA4</i>, and <i>VCX</i> family genes-may contribute to the pathogenesis of neurological and cardiac abnormalities, warranting further functional studies and long-term clinical monitoring.</p>\",\"PeriodicalId\":23294,\"journal\":{\"name\":\"Translational pediatrics\",\"volume\":\"14 6\",\"pages\":\"1370-1379\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12268512/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational pediatrics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tp-2025-87\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/6/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tp-2025-87","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
Cardiac arrhythmia, developmental delay, epilepsy and ichthyosis due to Xp22.31 deletion: review of literature and case report.
Background: The broad phenotypic variability observed in patients with Xp22.31 deletion, traditionally associated with X-linked ichthyosis (XLI), is increasingly recognized as encompassing a wider spectrum of clinical manifestations. While ichthyosis, caused by STS gene deletion, remains the hallmark feature, recent studies and genomic analyses (e.g., chromosomal microarray and whole genome sequencing) have revealed additional extracutaneous phenotypes. These include corneal opacification, cryptorchidism, autism spectrum disorders, intellectual disability, epilepsy, developmental delay, renal anomalies, and an elevated risk of atrial fibrillation and other cardiac arrhythmias, particularly in males. Interestingly, duplications of this region are usually considered benign, underscoring the need for nuanced interpretation.
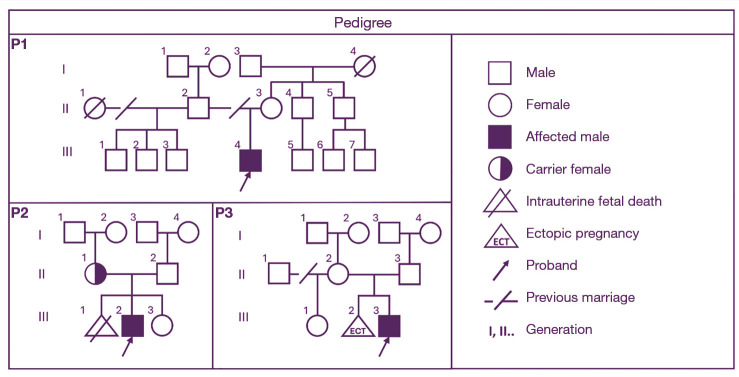
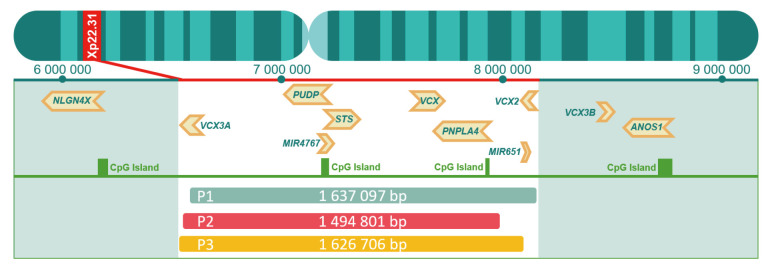
Case description: We describe three unrelated male patients carrying hemizygous Xp22.31 microdeletions (~1.6 Mb), all presenting with mild to moderate ichthyosis characterized by "plate-like" desquamation. Two patients exhibited intellectual disability and bradyarrhythmia, while one experienced seizures. None had major congenital anomalies, and all underwent chromosomal microarray analysis to confirm the diagnosis.
Conclusions: Our findings emphasize the need for a multidisciplinary approach when evaluating patients with Xp22.31 deletions, extending beyond dermatologic assessment to include neurological and cardiological evaluations, even in the absence of overt symptoms. This broader phenotypic understanding may enhance clinical management, support more accurate genetic counseling, and inform prenatal diagnostic decision-making. Furthermore, our observations support the hypothesis that genes within the deleted region-such as STS, PNPLA4, and VCX family genes-may contribute to the pathogenesis of neurological and cardiac abnormalities, warranting further functional studies and long-term clinical monitoring.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


