Harindi R Atapattu, Sahar Bayat, Henry Pruett, Anton S Perera, Tareq Hossain, Keerthan R. Rao, Kevin Pedersen, Augustine Yusuf, Sean Parkin, Chad Risko and Kenneth R. Graham*,
{"title":"层状金属卤化物钙钛矿的A′位偶极子大小和方向决定电离能和电子亲和力。","authors":"Harindi R Atapattu, Sahar Bayat, Henry Pruett, Anton S Perera, Tareq Hossain, Keerthan R. Rao, Kevin Pedersen, Augustine Yusuf, Sean Parkin, Chad Risko and Kenneth R. Graham*, ","doi":"10.1021/jacs.5c08621","DOIUrl":null,"url":null,"abstract":"<p >Layered metal halide perovskites (LHPs), often referred to as 2D HPs, are promising materials for developing optoelectronics due to their tunable optoelectronic properties and improved stability compared to nonlayered (3D) metal halide perovskites. For integration into electronic devices, it is critical to appropriately adjust the work function (WF) and transport energies of the LHPs to promote efficient charge transfer between materials in the device stack. The transport energies of LHPs can be modified by changing the A’-site cation structure, inorganic sheet thickness, and the metal cation or halide anion. Here, we investigate how the A’-site cation structure influences the WF, ionization energy (IE), and electron affinity (EA) of <i>n</i> = 1 Sn- and Pb-based LHPs with a series of <i>ortho-</i> and <i>para</i>-functionalized phenethylammonium (PEA) iodide derivatives. To accurately assign the IE and EA, we develop a fitting method where the instrumental broadening, σ<sub>IB</sub>, in ultraviolet and low-energy inverse photoemission spectroscopy (UPS and LEIPS, respectively) is accounted for. Density functional theory calculations combined with UPS and LEIPS measurements show that the dipole magnitude and direction of the A’-site cation exert a dominant influence on the WF, IE, and EA. Here, the direction and magnitude of the dipole, as manipulated through the strength and position of the electron-withdrawing or -donating substituent on PEA, can tune the WF by up to 1.2 eV, the IE by up to 0.9 eV, and the EA by up to 1.2 eV. The crystal structures indicate that the Sn–I–Sn bond angles have a clear influence over the optical gap; however, the influence of these Sn–I–Sn bond angles on the transport energies is dwarfed by the effect of the A’ dipole. These results provide insight into how to tune the WF and transport energies of LHPs for optoelectronic device integration.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"147 30","pages":"26898–26906"},"PeriodicalIF":15.6000,"publicationDate":"2025-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A’-Site Dipole Magnitude and Direction Dominate the Ionization Energy and Electron Affinity of Layered Metal-Halide Perovskites\",\"authors\":\"Harindi R Atapattu, Sahar Bayat, Henry Pruett, Anton S Perera, Tareq Hossain, Keerthan R. Rao, Kevin Pedersen, Augustine Yusuf, Sean Parkin, Chad Risko and Kenneth R. Graham*, \",\"doi\":\"10.1021/jacs.5c08621\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Layered metal halide perovskites (LHPs), often referred to as 2D HPs, are promising materials for developing optoelectronics due to their tunable optoelectronic properties and improved stability compared to nonlayered (3D) metal halide perovskites. For integration into electronic devices, it is critical to appropriately adjust the work function (WF) and transport energies of the LHPs to promote efficient charge transfer between materials in the device stack. The transport energies of LHPs can be modified by changing the A’-site cation structure, inorganic sheet thickness, and the metal cation or halide anion. Here, we investigate how the A’-site cation structure influences the WF, ionization energy (IE), and electron affinity (EA) of <i>n</i> = 1 Sn- and Pb-based LHPs with a series of <i>ortho-</i> and <i>para</i>-functionalized phenethylammonium (PEA) iodide derivatives. To accurately assign the IE and EA, we develop a fitting method where the instrumental broadening, σ<sub>IB</sub>, in ultraviolet and low-energy inverse photoemission spectroscopy (UPS and LEIPS, respectively) is accounted for. Density functional theory calculations combined with UPS and LEIPS measurements show that the dipole magnitude and direction of the A’-site cation exert a dominant influence on the WF, IE, and EA. Here, the direction and magnitude of the dipole, as manipulated through the strength and position of the electron-withdrawing or -donating substituent on PEA, can tune the WF by up to 1.2 eV, the IE by up to 0.9 eV, and the EA by up to 1.2 eV. The crystal structures indicate that the Sn–I–Sn bond angles have a clear influence over the optical gap; however, the influence of these Sn–I–Sn bond angles on the transport energies is dwarfed by the effect of the A’ dipole. These results provide insight into how to tune the WF and transport energies of LHPs for optoelectronic device integration.</p>\",\"PeriodicalId\":49,\"journal\":{\"name\":\"Journal of the American Chemical Society\",\"volume\":\"147 30\",\"pages\":\"26898–26906\"},\"PeriodicalIF\":15.6000,\"publicationDate\":\"2025-07-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jacs.5c08621\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.5c08621","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
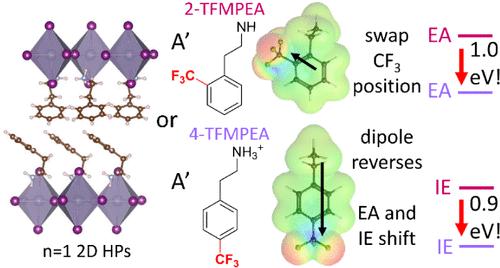
A’-Site Dipole Magnitude and Direction Dominate the Ionization Energy and Electron Affinity of Layered Metal-Halide Perovskites
Layered metal halide perovskites (LHPs), often referred to as 2D HPs, are promising materials for developing optoelectronics due to their tunable optoelectronic properties and improved stability compared to nonlayered (3D) metal halide perovskites. For integration into electronic devices, it is critical to appropriately adjust the work function (WF) and transport energies of the LHPs to promote efficient charge transfer between materials in the device stack. The transport energies of LHPs can be modified by changing the A’-site cation structure, inorganic sheet thickness, and the metal cation or halide anion. Here, we investigate how the A’-site cation structure influences the WF, ionization energy (IE), and electron affinity (EA) of n = 1 Sn- and Pb-based LHPs with a series of ortho- and para-functionalized phenethylammonium (PEA) iodide derivatives. To accurately assign the IE and EA, we develop a fitting method where the instrumental broadening, σIB, in ultraviolet and low-energy inverse photoemission spectroscopy (UPS and LEIPS, respectively) is accounted for. Density functional theory calculations combined with UPS and LEIPS measurements show that the dipole magnitude and direction of the A’-site cation exert a dominant influence on the WF, IE, and EA. Here, the direction and magnitude of the dipole, as manipulated through the strength and position of the electron-withdrawing or -donating substituent on PEA, can tune the WF by up to 1.2 eV, the IE by up to 0.9 eV, and the EA by up to 1.2 eV. The crystal structures indicate that the Sn–I–Sn bond angles have a clear influence over the optical gap; however, the influence of these Sn–I–Sn bond angles on the transport energies is dwarfed by the effect of the A’ dipole. These results provide insight into how to tune the WF and transport energies of LHPs for optoelectronic device integration.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


