Francisco Fuentes-Santander, Carolina Curiqueo, Rafael Araos, Juan A Ugalde
{"title":"BugBuster:一种用于宏基因组数据分析的新颖的自动和可重复的工作流程。","authors":"Francisco Fuentes-Santander, Carolina Curiqueo, Rafael Araos, Juan A Ugalde","doi":"10.1093/bioadv/vbaf152","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>Metagenomic sequencing generates massive datasets that capture the complete genetic content of a sample, enabling detailed characterization of microbial communities. Yet the software and processes necessary to transform raw data into biologically meaningful results have become increasingly complex, limiting accessibility for researchers without specialist expertise. In this work, we present a novel modular a reproducible workflow developed to facilitate the analysis of metagenomic data. BugBuster is a fully containerized, modular, and reproducible workflow implemented in Nextflow. The pipeline streamlines analysis at level of reads, contigs, and metagenome-assembled genomes, offering dedicated modules for taxonomic profiling and resistome characterization. Thanks to the use of containers, BugBuster can be deployed with minimal configuration on workstations, high-performance clusters, or cloud platforms. Together, these features allow the robust, scalable, and reproducible analysis of metagenomic datasets.</p><p><strong>Availability and implementation: </strong>BugBuster was written in Nextflow-DSL2. The program applications, user manual, example data and code are freely available at https://github.com/gene2dis/BugBuster.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf152"},"PeriodicalIF":2.8000,"publicationDate":"2025-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12255886/pdf/","citationCount":"0","resultStr":"{\"title\":\"BugBuster: a novel automatic and reproducible workflow for metagenomic data analysis.\",\"authors\":\"Francisco Fuentes-Santander, Carolina Curiqueo, Rafael Araos, Juan A Ugalde\",\"doi\":\"10.1093/bioadv/vbaf152\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Summary: </strong>Metagenomic sequencing generates massive datasets that capture the complete genetic content of a sample, enabling detailed characterization of microbial communities. Yet the software and processes necessary to transform raw data into biologically meaningful results have become increasingly complex, limiting accessibility for researchers without specialist expertise. In this work, we present a novel modular a reproducible workflow developed to facilitate the analysis of metagenomic data. BugBuster is a fully containerized, modular, and reproducible workflow implemented in Nextflow. The pipeline streamlines analysis at level of reads, contigs, and metagenome-assembled genomes, offering dedicated modules for taxonomic profiling and resistome characterization. Thanks to the use of containers, BugBuster can be deployed with minimal configuration on workstations, high-performance clusters, or cloud platforms. Together, these features allow the robust, scalable, and reproducible analysis of metagenomic datasets.</p><p><strong>Availability and implementation: </strong>BugBuster was written in Nextflow-DSL2. The program applications, user manual, example data and code are freely available at https://github.com/gene2dis/BugBuster.</p>\",\"PeriodicalId\":72368,\"journal\":{\"name\":\"Bioinformatics advances\",\"volume\":\"5 1\",\"pages\":\"vbaf152\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-06-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12255886/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioadv/vbaf152\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf152","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
BugBuster: a novel automatic and reproducible workflow for metagenomic data analysis.
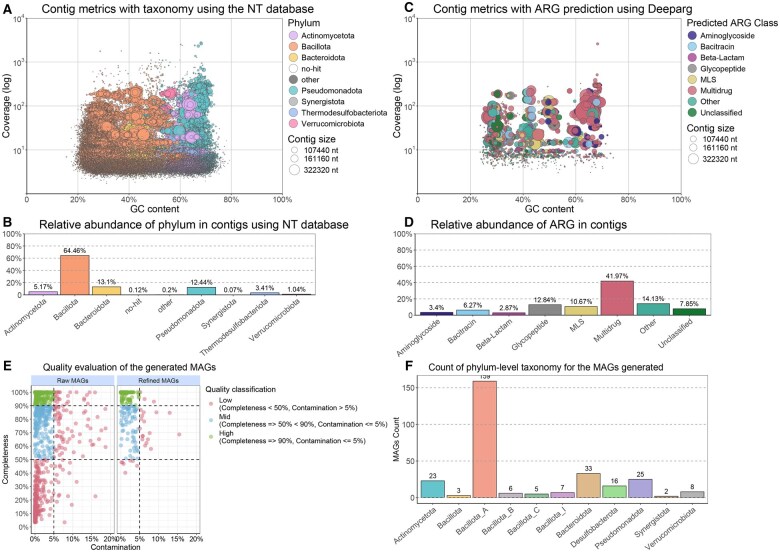
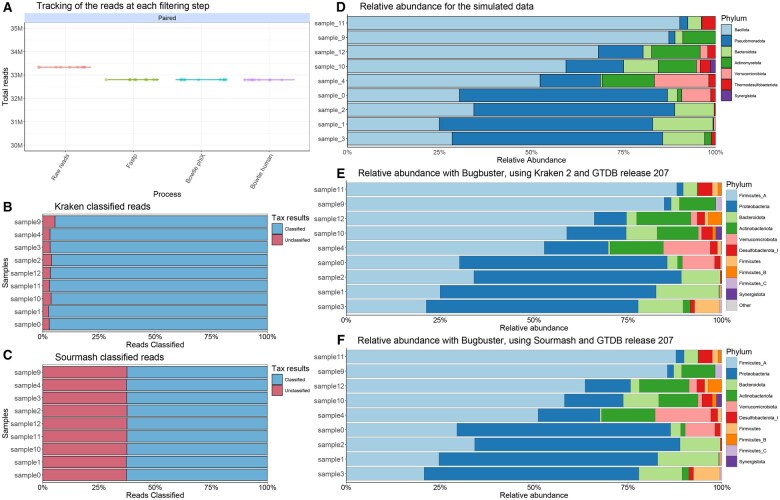
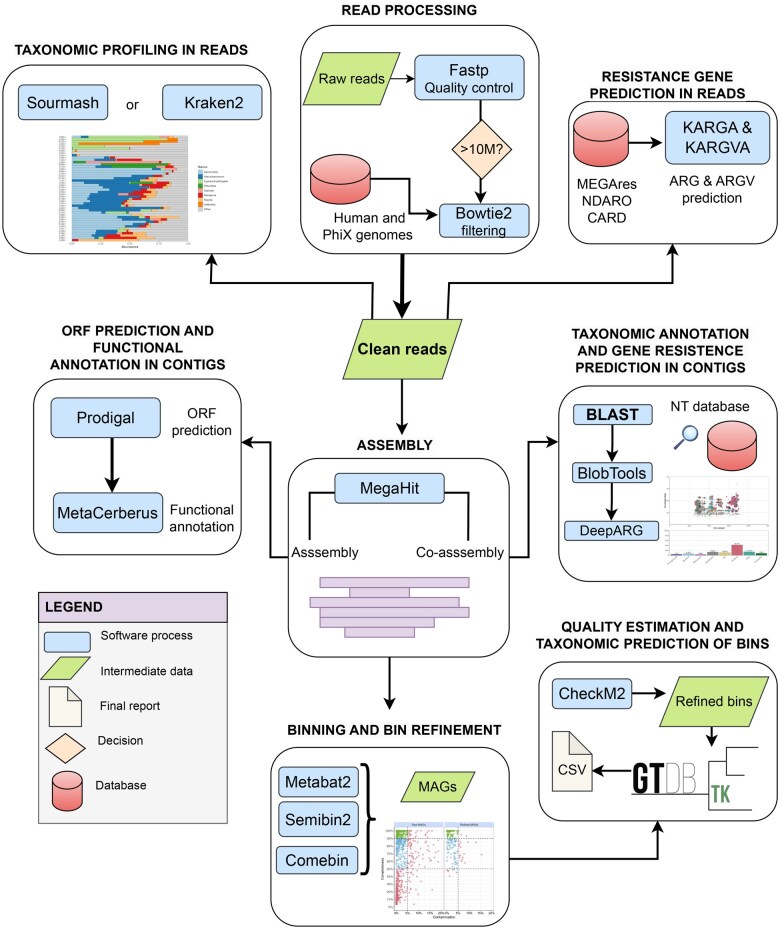
Summary: Metagenomic sequencing generates massive datasets that capture the complete genetic content of a sample, enabling detailed characterization of microbial communities. Yet the software and processes necessary to transform raw data into biologically meaningful results have become increasingly complex, limiting accessibility for researchers without specialist expertise. In this work, we present a novel modular a reproducible workflow developed to facilitate the analysis of metagenomic data. BugBuster is a fully containerized, modular, and reproducible workflow implemented in Nextflow. The pipeline streamlines analysis at level of reads, contigs, and metagenome-assembled genomes, offering dedicated modules for taxonomic profiling and resistome characterization. Thanks to the use of containers, BugBuster can be deployed with minimal configuration on workstations, high-performance clusters, or cloud platforms. Together, these features allow the robust, scalable, and reproducible analysis of metagenomic datasets.
Availability and implementation: BugBuster was written in Nextflow-DSL2. The program applications, user manual, example data and code are freely available at https://github.com/gene2dis/BugBuster.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


