Anton Cherednichenko, Sergii Afonin, Oleg Babii, Taras Voitsitskyi, Roman Stratiichuk, Ihor Koleiev, Volodymyr Vozniak, Nazar Shevchuk, Zakhar Ostrovsky, Semen Yesylevskyy, Alan Nafiiev, Serhii Starosyla, Anne S Ulrich, Aigars Jirgensons, Igor V Komarov
{"title":"利用分子动力学数据预测生物活性的神经网络模型:以光开关肽为例。","authors":"Anton Cherednichenko, Sergii Afonin, Oleg Babii, Taras Voitsitskyi, Roman Stratiichuk, Ihor Koleiev, Volodymyr Vozniak, Nazar Shevchuk, Zakhar Ostrovsky, Semen Yesylevskyy, Alan Nafiiev, Serhii Starosyla, Anne S Ulrich, Aigars Jirgensons, Igor V Komarov","doi":"10.1002/minf.70001","DOIUrl":null,"url":null,"abstract":"<p><p>Prediction of biological activities of chemical compounds by the machine learning techniques in general and the neural networks (NNs) in particular, is usually based on the analysis of their binding to the target of interest. If such affinity data is not available, the ligand-based approaches can be used where the NN models are trained to assess similarity of compounds to those with known biological activity. Obviously, this approach only works well if the similarity between the training set and the evaluated molecules is sufficiently high. In the case of large and conformationally flexible organic compounds, the activity becomes dependent not only on chemical identity but also on the dynamics of molecular motions, which imposes significant challenges to existing approaches based on static structural 2D and 3D molecular descriptors. A prominent example of compounds, which are especially challenging for existing NN activity prediction techniques, are photoswitchable macrocyclic peptides containing a diarylethene \"photoswitch\" (DAE). These molecules exist in two isomeric forms with remarkably different biological activities, which are interconvertible by light of different wavelengths. Activity prediction models have to distinguish in this case not only between the different peptides but also between the photoisomers of the same peptide. In this work, we demonstrate that the features extracted from classical molecular dynamics (MD) trajectories are superior to conventional 2D or 3D descriptor-based features when used in activity prediction NN models of DAE-containing photoswitchable peptides. Using MD-derived features, we successfully created two NN models that predict activities of photoswitchable peptidomimetics, analogs of the natural peptidic antibiotic gramicidin S. The first model precisely predicts the cytotoxic activity of similar peptide analogs. The second model reliably predicts the differences in the biological activities of DAE photoisomers of the same peptide, even if the type of its activity differs from one in the training dataset. Our results demonstrate that accounting for MD-derived dynamic features allows generalizing the ligand-based activity prediction NN models to the cases of large and conformationally flexible molecules, which were previously considered intractable by this class of models.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"44 7","pages":"e70001"},"PeriodicalIF":3.1000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12257427/pdf/","citationCount":"0","resultStr":"{\"title\":\"Neural Network Models for Prediction of Biological Activity using Molecular Dynamics Data: A Case of Photoswitchable Peptides.\",\"authors\":\"Anton Cherednichenko, Sergii Afonin, Oleg Babii, Taras Voitsitskyi, Roman Stratiichuk, Ihor Koleiev, Volodymyr Vozniak, Nazar Shevchuk, Zakhar Ostrovsky, Semen Yesylevskyy, Alan Nafiiev, Serhii Starosyla, Anne S Ulrich, Aigars Jirgensons, Igor V Komarov\",\"doi\":\"10.1002/minf.70001\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Prediction of biological activities of chemical compounds by the machine learning techniques in general and the neural networks (NNs) in particular, is usually based on the analysis of their binding to the target of interest. If such affinity data is not available, the ligand-based approaches can be used where the NN models are trained to assess similarity of compounds to those with known biological activity. Obviously, this approach only works well if the similarity between the training set and the evaluated molecules is sufficiently high. In the case of large and conformationally flexible organic compounds, the activity becomes dependent not only on chemical identity but also on the dynamics of molecular motions, which imposes significant challenges to existing approaches based on static structural 2D and 3D molecular descriptors. A prominent example of compounds, which are especially challenging for existing NN activity prediction techniques, are photoswitchable macrocyclic peptides containing a diarylethene \\\"photoswitch\\\" (DAE). These molecules exist in two isomeric forms with remarkably different biological activities, which are interconvertible by light of different wavelengths. Activity prediction models have to distinguish in this case not only between the different peptides but also between the photoisomers of the same peptide. In this work, we demonstrate that the features extracted from classical molecular dynamics (MD) trajectories are superior to conventional 2D or 3D descriptor-based features when used in activity prediction NN models of DAE-containing photoswitchable peptides. Using MD-derived features, we successfully created two NN models that predict activities of photoswitchable peptidomimetics, analogs of the natural peptidic antibiotic gramicidin S. The first model precisely predicts the cytotoxic activity of similar peptide analogs. The second model reliably predicts the differences in the biological activities of DAE photoisomers of the same peptide, even if the type of its activity differs from one in the training dataset. Our results demonstrate that accounting for MD-derived dynamic features allows generalizing the ligand-based activity prediction NN models to the cases of large and conformationally flexible molecules, which were previously considered intractable by this class of models.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\"44 7\",\"pages\":\"e70001\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12257427/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.70001\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.70001","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Neural Network Models for Prediction of Biological Activity using Molecular Dynamics Data: A Case of Photoswitchable Peptides.
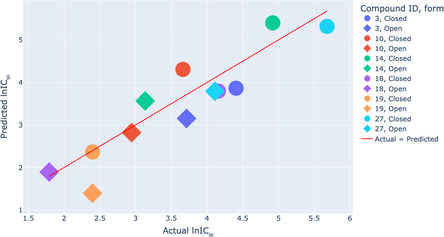
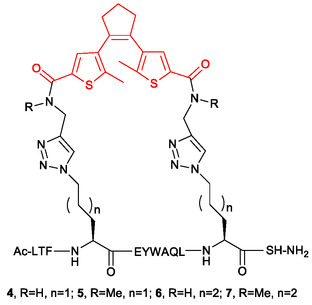
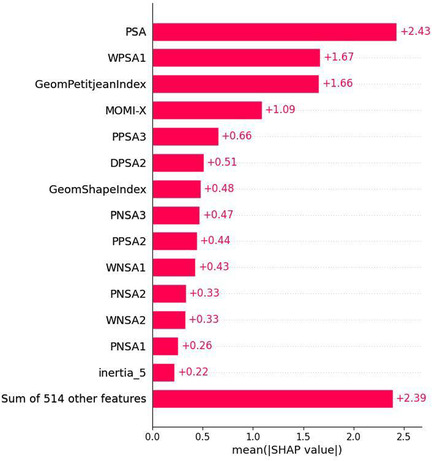
Prediction of biological activities of chemical compounds by the machine learning techniques in general and the neural networks (NNs) in particular, is usually based on the analysis of their binding to the target of interest. If such affinity data is not available, the ligand-based approaches can be used where the NN models are trained to assess similarity of compounds to those with known biological activity. Obviously, this approach only works well if the similarity between the training set and the evaluated molecules is sufficiently high. In the case of large and conformationally flexible organic compounds, the activity becomes dependent not only on chemical identity but also on the dynamics of molecular motions, which imposes significant challenges to existing approaches based on static structural 2D and 3D molecular descriptors. A prominent example of compounds, which are especially challenging for existing NN activity prediction techniques, are photoswitchable macrocyclic peptides containing a diarylethene "photoswitch" (DAE). These molecules exist in two isomeric forms with remarkably different biological activities, which are interconvertible by light of different wavelengths. Activity prediction models have to distinguish in this case not only between the different peptides but also between the photoisomers of the same peptide. In this work, we demonstrate that the features extracted from classical molecular dynamics (MD) trajectories are superior to conventional 2D or 3D descriptor-based features when used in activity prediction NN models of DAE-containing photoswitchable peptides. Using MD-derived features, we successfully created two NN models that predict activities of photoswitchable peptidomimetics, analogs of the natural peptidic antibiotic gramicidin S. The first model precisely predicts the cytotoxic activity of similar peptide analogs. The second model reliably predicts the differences in the biological activities of DAE photoisomers of the same peptide, even if the type of its activity differs from one in the training dataset. Our results demonstrate that accounting for MD-derived dynamic features allows generalizing the ligand-based activity prediction NN models to the cases of large and conformationally flexible molecules, which were previously considered intractable by this class of models.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


