Robert C. Glen, Jason C. Cole, James J. P. Stewart
{"title":"结合蛋白质-配体对接和半经验量子力学预测酶抑制(IC50)。","authors":"Robert C. Glen, Jason C. Cole, James J. P. Stewart","doi":"10.1007/s00894-025-06423-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The ability to predict the relative binding energies of ligands to a biological receptor would be of great value in drug discovery. However, accurately calculating the predicted binding energies is limited by the high accuracy required, by the presence of multiple minima on the potential energy surface, and by issues specific to the intrinsic properties of the binding site, such as details of the geometry of the ligand–protein complex. To address these issues, a systematic analysis of potential sources of error was carried out which resulted in a few relatively small changes being made to the MOPAC program.</p><h3>Methods</h3><p>A set of 77 ligands was constructed for which experimentally determined IC<sub>50</sub> values were available. For each of the ligands, prediction of the protein–ligand interaction energy was carried out in two distinct stages. In the first stage, the Protein–Ligand docking program GOLD was used to generate several distinct conformations of the ligand bound to a protein. The geometries of these systems were then optimised using the MOPAC program. A comparison of the relative binding energies of the ligands with the reported IC<sub>50</sub> values showed a very poor predictive power. By partitioning the ligand set into two subsets, and eliminating six ligands that were inconsistent with the experimental results, a large increase in accuracy was obtained.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12255584/pdf/","citationCount":"0","resultStr":"{\"title\":\"Prediction of enzyme inhibition (IC50) using a combination of protein–ligand docking and semiempirical quantum mechanics\",\"authors\":\"Robert C. Glen, Jason C. Cole, James J. P. Stewart\",\"doi\":\"10.1007/s00894-025-06423-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>The ability to predict the relative binding energies of ligands to a biological receptor would be of great value in drug discovery. However, accurately calculating the predicted binding energies is limited by the high accuracy required, by the presence of multiple minima on the potential energy surface, and by issues specific to the intrinsic properties of the binding site, such as details of the geometry of the ligand–protein complex. To address these issues, a systematic analysis of potential sources of error was carried out which resulted in a few relatively small changes being made to the MOPAC program.</p><h3>Methods</h3><p>A set of 77 ligands was constructed for which experimentally determined IC<sub>50</sub> values were available. For each of the ligands, prediction of the protein–ligand interaction energy was carried out in two distinct stages. In the first stage, the Protein–Ligand docking program GOLD was used to generate several distinct conformations of the ligand bound to a protein. The geometries of these systems were then optimised using the MOPAC program. A comparison of the relative binding energies of the ligands with the reported IC<sub>50</sub> values showed a very poor predictive power. By partitioning the ligand set into two subsets, and eliminating six ligands that were inconsistent with the experimental results, a large increase in accuracy was obtained.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 8\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-07-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12255584/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06423-7\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06423-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Prediction of enzyme inhibition (IC50) using a combination of protein–ligand docking and semiempirical quantum mechanics
Context
The ability to predict the relative binding energies of ligands to a biological receptor would be of great value in drug discovery. However, accurately calculating the predicted binding energies is limited by the high accuracy required, by the presence of multiple minima on the potential energy surface, and by issues specific to the intrinsic properties of the binding site, such as details of the geometry of the ligand–protein complex. To address these issues, a systematic analysis of potential sources of error was carried out which resulted in a few relatively small changes being made to the MOPAC program.
Methods
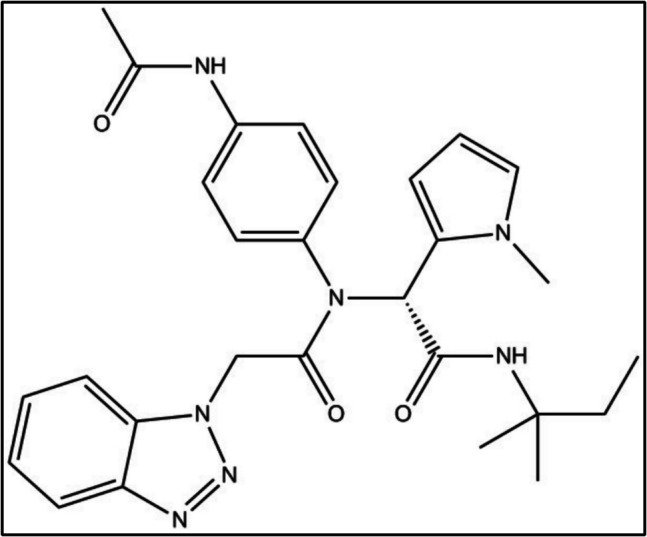
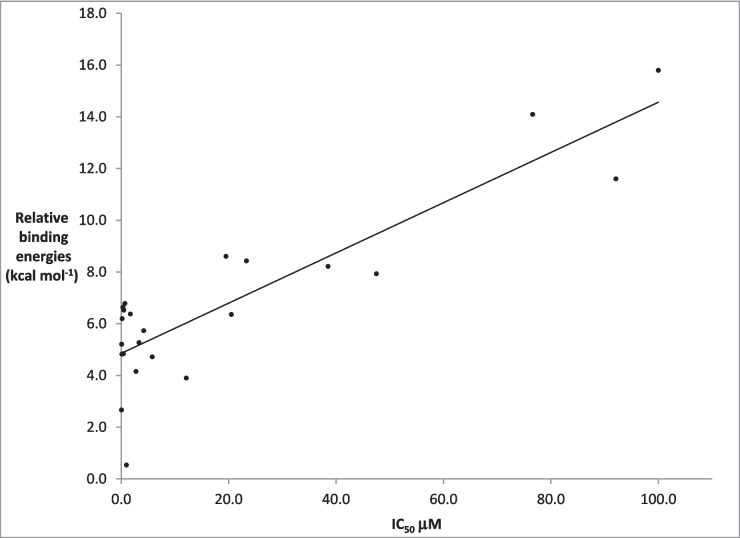
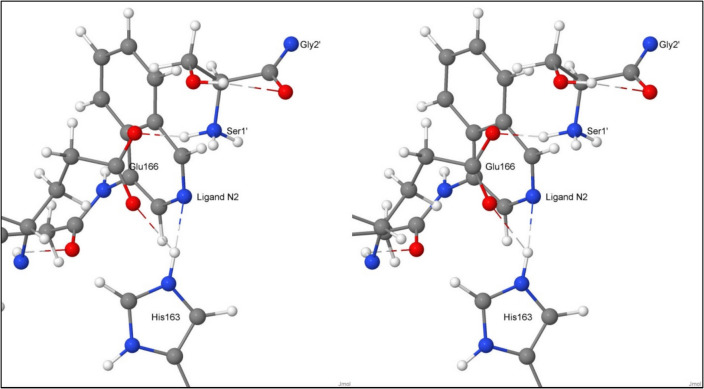
A set of 77 ligands was constructed for which experimentally determined IC50 values were available. For each of the ligands, prediction of the protein–ligand interaction energy was carried out in two distinct stages. In the first stage, the Protein–Ligand docking program GOLD was used to generate several distinct conformations of the ligand bound to a protein. The geometries of these systems were then optimised using the MOPAC program. A comparison of the relative binding energies of the ligands with the reported IC50 values showed a very poor predictive power. By partitioning the ligand set into two subsets, and eliminating six ligands that were inconsistent with the experimental results, a large increase in accuracy was obtained.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


