{"title":"镍催化1,3-二烯与甲醇区域选择性和对映选择性氢烷氧基化的DFT研究:内球与外球机制","authors":"Xia Zhao, Yong Xia and Xiaotai Wang","doi":"10.1039/D5QO00814J","DOIUrl":null,"url":null,"abstract":"<p >We present a density functional theory (DFT) study on a remarkable nickel-catalyzed regio- and enantioselective hydroalkoxylation and C–O bond formation reaction of 1,3-dienes with methanol (a common yet underutilized oxygen nucleophile). An in-depth computational analysis, aided by the Curtin–Hammett principle, unravels this complex reaction and provides novel mechanistic insights. The catalytically active species, a π complex with the diene substrate, undergoes a regioselective proton transfer from methanol to the diene. This ligand-to-ligand hydrogen transfer (LLHT) proceeds <em>via</em> an outer-sphere mechanism, critically facilitated by additional H-bonded methanol molecules, rather than the originally proposed inner-sphere mechanism without hydrogen-bonding assistance. The regioselectivity of the LLHT step is governed by the electronic effects and hydrogen bonding interactions. Subsequently, the resulting nickel η<small><sup>3</sup></small>-allyl intermediate initiates an enantioselective outer-sphere nucleophilic attack by the methoxide anion on the allyl ligand, yielding the chiral product. The origins of the enantioselectivity include stronger noncovalent C(sp<small><sup>3</sup></small>)–H/π interactions and the absence of steric hindrance in the key transition state that leads to the (<em>S</em>)-enantiomer. The mechanistic insights uncovered in this study can guide the further development of nickel-catalyzed C–O bond-forming reactions utilizing alkenes and alcohols as substrates.</p>","PeriodicalId":97,"journal":{"name":"Organic Chemistry Frontiers","volume":" 21","pages":" 5938-5946"},"PeriodicalIF":4.7000,"publicationDate":"2025-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DFT study of nickel-catalyzed regio- and enantioselective hydroalkoxylation of 1,3-dienes with methanol: inner-sphere versus outer-sphere mechanisms†\",\"authors\":\"Xia Zhao, Yong Xia and Xiaotai Wang\",\"doi\":\"10.1039/D5QO00814J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present a density functional theory (DFT) study on a remarkable nickel-catalyzed regio- and enantioselective hydroalkoxylation and C–O bond formation reaction of 1,3-dienes with methanol (a common yet underutilized oxygen nucleophile). An in-depth computational analysis, aided by the Curtin–Hammett principle, unravels this complex reaction and provides novel mechanistic insights. The catalytically active species, a π complex with the diene substrate, undergoes a regioselective proton transfer from methanol to the diene. This ligand-to-ligand hydrogen transfer (LLHT) proceeds <em>via</em> an outer-sphere mechanism, critically facilitated by additional H-bonded methanol molecules, rather than the originally proposed inner-sphere mechanism without hydrogen-bonding assistance. The regioselectivity of the LLHT step is governed by the electronic effects and hydrogen bonding interactions. Subsequently, the resulting nickel η<small><sup>3</sup></small>-allyl intermediate initiates an enantioselective outer-sphere nucleophilic attack by the methoxide anion on the allyl ligand, yielding the chiral product. The origins of the enantioselectivity include stronger noncovalent C(sp<small><sup>3</sup></small>)–H/π interactions and the absence of steric hindrance in the key transition state that leads to the (<em>S</em>)-enantiomer. The mechanistic insights uncovered in this study can guide the further development of nickel-catalyzed C–O bond-forming reactions utilizing alkenes and alcohols as substrates.</p>\",\"PeriodicalId\":97,\"journal\":{\"name\":\"Organic Chemistry Frontiers\",\"volume\":\" 21\",\"pages\":\" 5938-5946\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2025-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Organic Chemistry Frontiers\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/qo/d5qo00814j\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organic Chemistry Frontiers","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/qo/d5qo00814j","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
DFT study of nickel-catalyzed regio- and enantioselective hydroalkoxylation of 1,3-dienes with methanol: inner-sphere versus outer-sphere mechanisms†
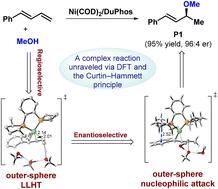
We present a density functional theory (DFT) study on a remarkable nickel-catalyzed regio- and enantioselective hydroalkoxylation and C–O bond formation reaction of 1,3-dienes with methanol (a common yet underutilized oxygen nucleophile). An in-depth computational analysis, aided by the Curtin–Hammett principle, unravels this complex reaction and provides novel mechanistic insights. The catalytically active species, a π complex with the diene substrate, undergoes a regioselective proton transfer from methanol to the diene. This ligand-to-ligand hydrogen transfer (LLHT) proceeds via an outer-sphere mechanism, critically facilitated by additional H-bonded methanol molecules, rather than the originally proposed inner-sphere mechanism without hydrogen-bonding assistance. The regioselectivity of the LLHT step is governed by the electronic effects and hydrogen bonding interactions. Subsequently, the resulting nickel η3-allyl intermediate initiates an enantioselective outer-sphere nucleophilic attack by the methoxide anion on the allyl ligand, yielding the chiral product. The origins of the enantioselectivity include stronger noncovalent C(sp3)–H/π interactions and the absence of steric hindrance in the key transition state that leads to the (S)-enantiomer. The mechanistic insights uncovered in this study can guide the further development of nickel-catalyzed C–O bond-forming reactions utilizing alkenes and alcohols as substrates.
期刊介绍:
Organic Chemistry Frontiers is an esteemed journal that publishes high-quality research across the field of organic chemistry. It places a significant emphasis on studies that contribute substantially to the field by introducing new or significantly improved protocols and methodologies. The journal covers a wide array of topics which include, but are not limited to, organic synthesis, the development of synthetic methodologies, catalysis, natural products, functional organic materials, supramolecular and macromolecular chemistry, as well as physical and computational organic chemistry.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


