二芳烯酮中烯烃的反离子和溶剂控制选择性硼氢化加氢反应。
Organic chemistry frontiers : an international journal of organic chemistry
Pub Date : 2025-04-28
DOI:10.1039/d5qo00883b
引用次数: 0
摘要
硼氢化物被认为是在烯烃存在下酮选择性氢化的基准试剂,这是有机教科书中描述的反应。然而,相反的情况,即硼氢化物促进烯烃在酮存在下的加氢反应,却很少被描述。本研究表明,在标准的非催化反应条件下,使用一定量的金属硼氢化物(即NaBH4)后,二芳基烯酮中的烯烃官能团优先氢化为酮。对于二芳基烯酮,机理研究表明,适当的阳离子取代硼氢化物(从Li+到K+)和高共轭末端烯烃的特殊配置的组合有利于高度选择性的1,4 -氢化物加成,在室温下反应30分钟后,在没有任何催化剂或添加剂的帮助下,以高收率获得α -苄基取代的丙烯酮。对于反式二芳基烯酮(查尔酮),质子共溶剂从MeOH简单地转变为缺电子、缺空间阻碍的醇,就会触发烯烃基团的选择性加氢。这些结果否定了硼氢化物对烯酮的反应活性,并为使用普通硼氢化物试剂进行选择性烯烃加氢反应开辟了道路,在合成程序中具有潜在的应用前景。本文章由计算机程序翻译,如有差异,请以英文原文为准。
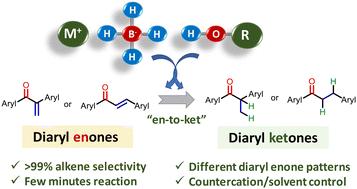
Countercation- and solvent-controlled selective borohydride hydrogenation of alkenes in diaryl enones†
Borohydrides are considered benchmark reagents for the selective hydrogenation of ketones in the presence of alkenes, a reaction described in organic textbooks. However, the opposite, i.e. the borohydride-promoted hydrogenation of an alkene in the presence of a ketone, is barely described. Here we show that the alkene functionality in diaryl enones is preferentially hydrogenated to the ketone under standard uncatalyzed reaction conditions, after using a stoichiometric amount of a metal borohydride (i.e. NaBH4). For gem-diaryl enones, mechanistic studies indicate that the combination of a suitably cation-substituted borohydride (from Li+ to K+) and the particular disposition of the highly-conjugated terminal alkene favors a highly selective 1,4-hydride addition, giving access to α-benzyl-substituted propiophenones in high yields, at room temperature and after just 30 min reaction time, without the assistance of any catalyst or additive. For trans-diaryl enones (chalcones), the simple change of the protic co-solvent from MeOH to electron-deficient and sterically-hindered alcohols triggers the selective hydrogenation of the alkene group. These results defy the established reactivity of borohydrides for enones and open a way to employ common borohydride reagents for selective alkene hydrogenation reactions, with potential application in synthetic chemistry.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
7.80
自引率
0.00%
发文量
0
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


