Elaheh Zarean, Shuai Li, Melissa C Southey, Pierre-Antoine Dugué
{"title":"肿瘤DNA甲基化用于乳腺癌亚型和预后预测的综述。","authors":"Elaheh Zarean, Shuai Li, Melissa C Southey, Pierre-Antoine Dugué","doi":"10.1186/s13148-025-01922-z","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation in breast tumours has been extensively studied and has provided valuable insights into the clinical heterogeneity of breast cancer. In this review, we summarise the current literature that has used DNA methylation markers to subtype breast cancer and predict progression and survival. Widespread methylation differences have been observed across breast cancer subtypes at both the candidate genes and in genome-wide analyses, most notably between oestrogen receptor (ER) positive and ER-negative subtypes and for triple-negative tumours. Studies that attempted to create breast cancer subtypes using methylation data showed limited agreement in their capacity to group breast tumours, possibly due to methodological differences. Although many studies have reported associations of tumour DNA methylation with breast cancer outcomes and used machine learning methods to derive prediction models for survival, the extent to which these would replicate in independent datasets is currently unclear. We conclude that despite the potential of genome-wide methylation markers to unravel the heterogeneity of breast cancer, they currently appear to have limited clinical utility. Larger studies and replication of findings across studies are required to address the limitations of the existing literature.</p>","PeriodicalId":10366,"journal":{"name":"Clinical Epigenetics","volume":"17 1","pages":"109"},"PeriodicalIF":4.4000,"publicationDate":"2025-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12220808/pdf/","citationCount":"0","resultStr":"{\"title\":\"A review of the use of tumour DNA methylation for breast cancer subtyping and prediction of outcomes.\",\"authors\":\"Elaheh Zarean, Shuai Li, Melissa C Southey, Pierre-Antoine Dugué\",\"doi\":\"10.1186/s13148-025-01922-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation in breast tumours has been extensively studied and has provided valuable insights into the clinical heterogeneity of breast cancer. In this review, we summarise the current literature that has used DNA methylation markers to subtype breast cancer and predict progression and survival. Widespread methylation differences have been observed across breast cancer subtypes at both the candidate genes and in genome-wide analyses, most notably between oestrogen receptor (ER) positive and ER-negative subtypes and for triple-negative tumours. Studies that attempted to create breast cancer subtypes using methylation data showed limited agreement in their capacity to group breast tumours, possibly due to methodological differences. Although many studies have reported associations of tumour DNA methylation with breast cancer outcomes and used machine learning methods to derive prediction models for survival, the extent to which these would replicate in independent datasets is currently unclear. We conclude that despite the potential of genome-wide methylation markers to unravel the heterogeneity of breast cancer, they currently appear to have limited clinical utility. Larger studies and replication of findings across studies are required to address the limitations of the existing literature.</p>\",\"PeriodicalId\":10366,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"17 1\",\"pages\":\"109\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-07-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12220808/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-025-01922-z\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-025-01922-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A review of the use of tumour DNA methylation for breast cancer subtyping and prediction of outcomes.
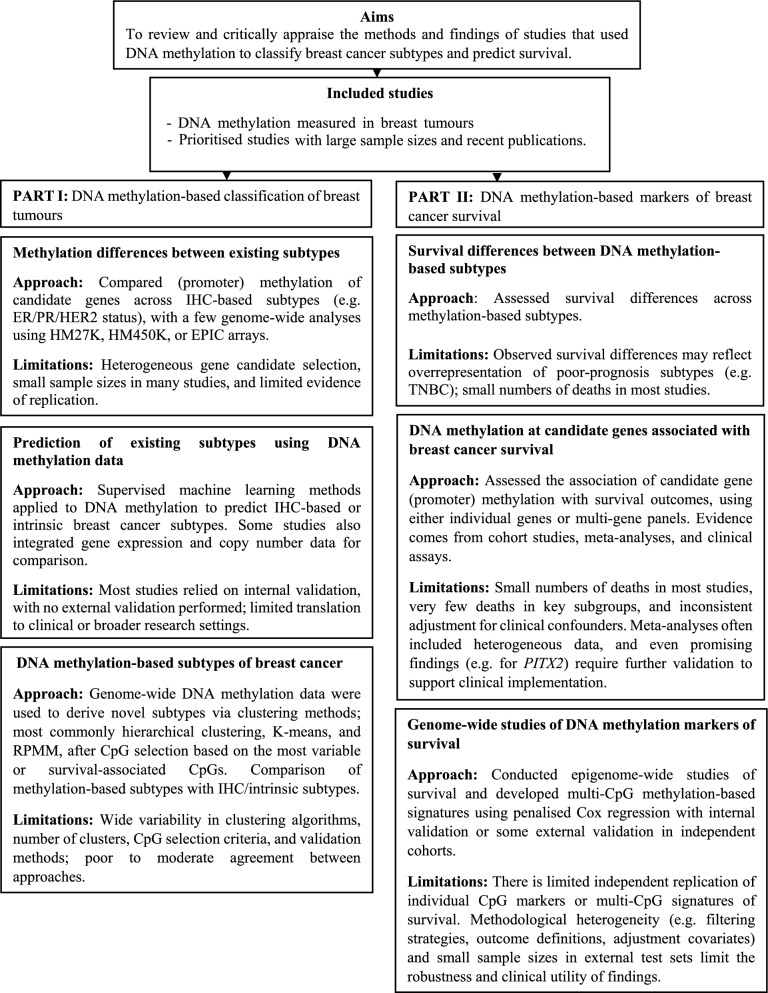
DNA methylation in breast tumours has been extensively studied and has provided valuable insights into the clinical heterogeneity of breast cancer. In this review, we summarise the current literature that has used DNA methylation markers to subtype breast cancer and predict progression and survival. Widespread methylation differences have been observed across breast cancer subtypes at both the candidate genes and in genome-wide analyses, most notably between oestrogen receptor (ER) positive and ER-negative subtypes and for triple-negative tumours. Studies that attempted to create breast cancer subtypes using methylation data showed limited agreement in their capacity to group breast tumours, possibly due to methodological differences. Although many studies have reported associations of tumour DNA methylation with breast cancer outcomes and used machine learning methods to derive prediction models for survival, the extent to which these would replicate in independent datasets is currently unclear. We conclude that despite the potential of genome-wide methylation markers to unravel the heterogeneity of breast cancer, they currently appear to have limited clinical utility. Larger studies and replication of findings across studies are required to address the limitations of the existing literature.
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


