Hanareia Ehau-Taumaunu, Terrence H Bell, Javad Sadeghi, Kevin L Hockett
{"title":"在实验微生物组选择后,番茄叶层抑制微生物组的快速和持续分化。","authors":"Hanareia Ehau-Taumaunu, Terrence H Bell, Javad Sadeghi, Kevin L Hockett","doi":"10.1186/s40793-025-00734-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Microbial-based treatments to protect plants against phytopathogens typically focus on soil-borne disease or the aboveground application of one or a few biocontrol microorganisms. However, diverse microbiomes may provide unique benefits to phytoprotection in the phyllosphere, by restricting pathogen access to niche space and/or through multiple forms of direct antagonism. We previously showed that successive experimental passaging of phyllosphere microbiomes along with the phytopathogen Pseudomonas syringae pv. tomato (Pto), which causes bacterial speck in tomato, led to the development of a disease suppressive microbial community. Here, we used amplicon sequencing to assess bacterial and fungal composition at the end of each passage, as well as shotgun metagenomics at key passages based on observed disease-suppressive phenotypes, to assess differences in functional potential between suppressive and non-suppressive communities.</p><p><strong>Results: </strong>Bacterial composition changed and diversity declined quickly due to passaging and remained low, particularly in treatments with Pto present, whereas fungal diversity did not. Pseudomonas and Xanthomonas populations were particularily enriched in disease-suppressive microbiomes compared to conducive microbiomes. The relative abundance of Pseudomonas syringae group gemonosp. 3 (the clade to which the introduced pathogen belongs) in shotgun metagenomic data was similar to what we observed for Pseudomonas ASVs in the 16S rRNA gene dataset. We also observed an increase in the abundance of genes associated with microbial antagonism at Passage 4, corresponding to the highest observed disease severity.</p><p><strong>Conclusions: </strong>Taxonomic richness and evenness were low within samples, with clustering occurring for suppressive or non-suppressive microbiomes. The relative abundance of genes associated with antagonism was higher for disease-suppressive phyllosphere microbiomes. This work is an important step towards understanding the microbe-microbe interactions within disease-suppressive phyllosphere communities.</p>","PeriodicalId":48553,"journal":{"name":"Environmental Microbiome","volume":"20 1","pages":"77"},"PeriodicalIF":5.4000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12211302/pdf/","citationCount":"0","resultStr":"{\"title\":\"Rapid and sustained differentiation of disease-suppressive phyllosphere microbiomes in tomato following experimental microbiome selection.\",\"authors\":\"Hanareia Ehau-Taumaunu, Terrence H Bell, Javad Sadeghi, Kevin L Hockett\",\"doi\":\"10.1186/s40793-025-00734-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Microbial-based treatments to protect plants against phytopathogens typically focus on soil-borne disease or the aboveground application of one or a few biocontrol microorganisms. However, diverse microbiomes may provide unique benefits to phytoprotection in the phyllosphere, by restricting pathogen access to niche space and/or through multiple forms of direct antagonism. We previously showed that successive experimental passaging of phyllosphere microbiomes along with the phytopathogen Pseudomonas syringae pv. tomato (Pto), which causes bacterial speck in tomato, led to the development of a disease suppressive microbial community. Here, we used amplicon sequencing to assess bacterial and fungal composition at the end of each passage, as well as shotgun metagenomics at key passages based on observed disease-suppressive phenotypes, to assess differences in functional potential between suppressive and non-suppressive communities.</p><p><strong>Results: </strong>Bacterial composition changed and diversity declined quickly due to passaging and remained low, particularly in treatments with Pto present, whereas fungal diversity did not. Pseudomonas and Xanthomonas populations were particularily enriched in disease-suppressive microbiomes compared to conducive microbiomes. The relative abundance of Pseudomonas syringae group gemonosp. 3 (the clade to which the introduced pathogen belongs) in shotgun metagenomic data was similar to what we observed for Pseudomonas ASVs in the 16S rRNA gene dataset. We also observed an increase in the abundance of genes associated with microbial antagonism at Passage 4, corresponding to the highest observed disease severity.</p><p><strong>Conclusions: </strong>Taxonomic richness and evenness were low within samples, with clustering occurring for suppressive or non-suppressive microbiomes. The relative abundance of genes associated with antagonism was higher for disease-suppressive phyllosphere microbiomes. This work is an important step towards understanding the microbe-microbe interactions within disease-suppressive phyllosphere communities.</p>\",\"PeriodicalId\":48553,\"journal\":{\"name\":\"Environmental Microbiome\",\"volume\":\"20 1\",\"pages\":\"77\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12211302/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Environmental Microbiome\",\"FirstCategoryId\":\"93\",\"ListUrlMain\":\"https://doi.org/10.1186/s40793-025-00734-1\",\"RegionNum\":2,\"RegionCategory\":\"环境科学与生态学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental Microbiome","FirstCategoryId":"93","ListUrlMain":"https://doi.org/10.1186/s40793-025-00734-1","RegionNum":2,"RegionCategory":"环境科学与生态学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Rapid and sustained differentiation of disease-suppressive phyllosphere microbiomes in tomato following experimental microbiome selection.
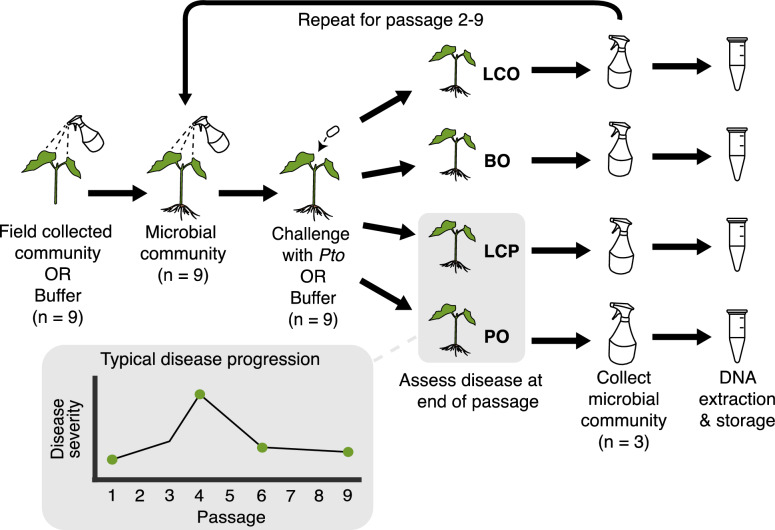
Background: Microbial-based treatments to protect plants against phytopathogens typically focus on soil-borne disease or the aboveground application of one or a few biocontrol microorganisms. However, diverse microbiomes may provide unique benefits to phytoprotection in the phyllosphere, by restricting pathogen access to niche space and/or through multiple forms of direct antagonism. We previously showed that successive experimental passaging of phyllosphere microbiomes along with the phytopathogen Pseudomonas syringae pv. tomato (Pto), which causes bacterial speck in tomato, led to the development of a disease suppressive microbial community. Here, we used amplicon sequencing to assess bacterial and fungal composition at the end of each passage, as well as shotgun metagenomics at key passages based on observed disease-suppressive phenotypes, to assess differences in functional potential between suppressive and non-suppressive communities.
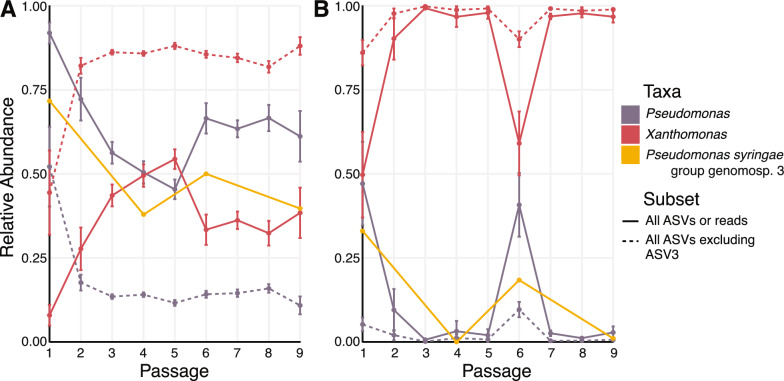
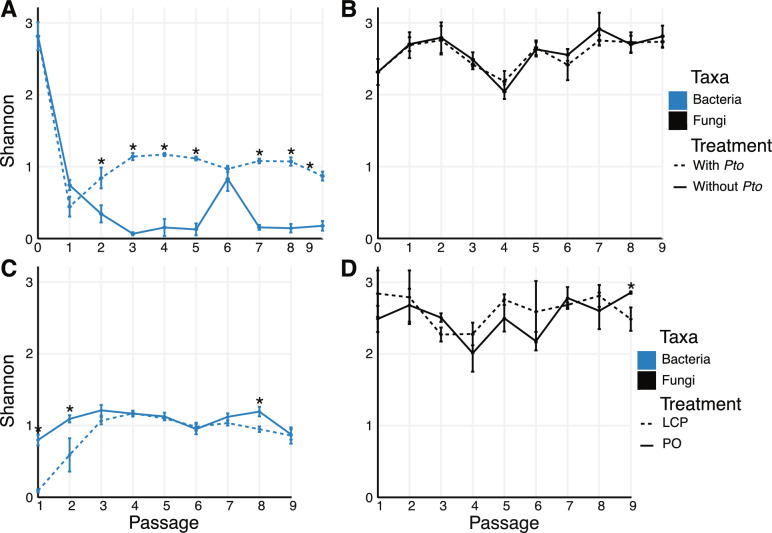
Results: Bacterial composition changed and diversity declined quickly due to passaging and remained low, particularly in treatments with Pto present, whereas fungal diversity did not. Pseudomonas and Xanthomonas populations were particularily enriched in disease-suppressive microbiomes compared to conducive microbiomes. The relative abundance of Pseudomonas syringae group gemonosp. 3 (the clade to which the introduced pathogen belongs) in shotgun metagenomic data was similar to what we observed for Pseudomonas ASVs in the 16S rRNA gene dataset. We also observed an increase in the abundance of genes associated with microbial antagonism at Passage 4, corresponding to the highest observed disease severity.
Conclusions: Taxonomic richness and evenness were low within samples, with clustering occurring for suppressive or non-suppressive microbiomes. The relative abundance of genes associated with antagonism was higher for disease-suppressive phyllosphere microbiomes. This work is an important step towards understanding the microbe-microbe interactions within disease-suppressive phyllosphere communities.
期刊介绍:
Microorganisms, omnipresent across Earth's diverse environments, play a crucial role in adapting to external changes, influencing Earth's systems and cycles, and contributing significantly to agricultural practices. Through applied microbiology, they offer solutions to various everyday needs. Environmental Microbiome recognizes the universal presence and significance of microorganisms, inviting submissions that explore the diverse facets of environmental and applied microbiological research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


