FuDong Wen, Yue Su, Dan Liu, YuPeng Wang, MeiNa Liu
{"title":"基于1位压缩感知和K-Medoids聚类的高维蛋白质组学数据自动稀疏特征选择。","authors":"FuDong Wen, Yue Su, Dan Liu, YuPeng Wang, MeiNa Liu","doi":"10.1186/s12859-025-06193-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>High-dimensional proteomics data present significant challenges in biomarker discovery due to technical noise, feature redundancy, and multicollinearity. Current feature selection methods, including filter, wrapper, and embedded approaches, struggle with stability, sparsity, and computational efficiency. To address these limitations, we propose Soft-Thresholded Compressed Sensing (ST-CS), a hybrid framework integrating 1-bit compressed sensing with K-Medoids clustering. Unlike conventional methods relying on manual thresholds, ST-CS automates feature selection by dynamically partitioning coefficient magnitudes into discriminative biomarkers and noise.</p><p><strong>Results: </strong>Evaluations on simulated and real-world proteomic datasets demonstrated ST-CS's superiority in feature selection capability and classification performance. In simulations, ST-CS achieved feature selection robustness with balanced sensitivity (> 80%) and specificity (> 99.8%), reducing false discovery rates (FDR) by 20-50% compared to Hard-Thresholded Compressed Sensing (HT-CS). Additionally, it attained superior F1 scores and Matthews Correlation Coefficients (MCC), outperforming HT-CS, LASSO, and SPLSDA in identifying true biomarkers while suppressing noise. For classification performance, ST-CS surpassed all methods in the area under the receiver operating characteristic curve (AUC) across varying noise levels while maintaining sparsity. Applied to Clinical Proteomic Tumor Analysis Consortium (CPTAC) datasets, ST-CS matched HT-CS's classification accuracy (AUC = 97.47% for intrahepatic cholangiocarcinoma) but with 57% fewer selected features (37 vs. 86), demonstrating its dual strength in precision biomarker discovery and predictive accuracy. For glioblastoma data, ST-CS achieved higher AUC (72.71%) than HT-CS (72.15%), LASSO (67.80%), and SPLSDA (71.38%) while retaining a parsimonious feature set (30 vs. 58 features for HT-CS). In ovarian serous cystadenocarcinoma, ST-CS further demonstrated its adaptability, attaining superior AUC (75.86%) over HT-CS (75.61%), LASSO (61.00%), and SPLSDA (70.75%) with only 24 ± 5 selected biomarkers. These results highlight ST-CS's ability to rigorously automate feature selection while balancing classification efficacy, interpretability, and scalability for translational proteomics.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"165"},"PeriodicalIF":3.3000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12220089/pdf/","citationCount":"0","resultStr":"{\"title\":\"Automated sparse feature selection in high-dimensional proteomics data via 1-bit compressed sensing and K-Medoids clustering.\",\"authors\":\"FuDong Wen, Yue Su, Dan Liu, YuPeng Wang, MeiNa Liu\",\"doi\":\"10.1186/s12859-025-06193-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>High-dimensional proteomics data present significant challenges in biomarker discovery due to technical noise, feature redundancy, and multicollinearity. Current feature selection methods, including filter, wrapper, and embedded approaches, struggle with stability, sparsity, and computational efficiency. To address these limitations, we propose Soft-Thresholded Compressed Sensing (ST-CS), a hybrid framework integrating 1-bit compressed sensing with K-Medoids clustering. Unlike conventional methods relying on manual thresholds, ST-CS automates feature selection by dynamically partitioning coefficient magnitudes into discriminative biomarkers and noise.</p><p><strong>Results: </strong>Evaluations on simulated and real-world proteomic datasets demonstrated ST-CS's superiority in feature selection capability and classification performance. In simulations, ST-CS achieved feature selection robustness with balanced sensitivity (> 80%) and specificity (> 99.8%), reducing false discovery rates (FDR) by 20-50% compared to Hard-Thresholded Compressed Sensing (HT-CS). Additionally, it attained superior F1 scores and Matthews Correlation Coefficients (MCC), outperforming HT-CS, LASSO, and SPLSDA in identifying true biomarkers while suppressing noise. For classification performance, ST-CS surpassed all methods in the area under the receiver operating characteristic curve (AUC) across varying noise levels while maintaining sparsity. Applied to Clinical Proteomic Tumor Analysis Consortium (CPTAC) datasets, ST-CS matched HT-CS's classification accuracy (AUC = 97.47% for intrahepatic cholangiocarcinoma) but with 57% fewer selected features (37 vs. 86), demonstrating its dual strength in precision biomarker discovery and predictive accuracy. For glioblastoma data, ST-CS achieved higher AUC (72.71%) than HT-CS (72.15%), LASSO (67.80%), and SPLSDA (71.38%) while retaining a parsimonious feature set (30 vs. 58 features for HT-CS). In ovarian serous cystadenocarcinoma, ST-CS further demonstrated its adaptability, attaining superior AUC (75.86%) over HT-CS (75.61%), LASSO (61.00%), and SPLSDA (70.75%) with only 24 ± 5 selected biomarkers. These results highlight ST-CS's ability to rigorously automate feature selection while balancing classification efficacy, interpretability, and scalability for translational proteomics.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"165\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12220089/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06193-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06193-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Automated sparse feature selection in high-dimensional proteomics data via 1-bit compressed sensing and K-Medoids clustering.
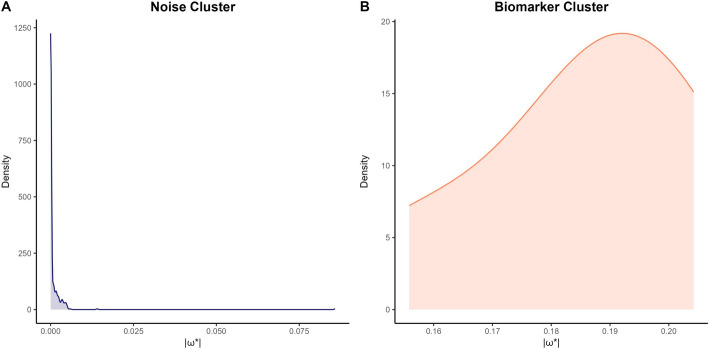
Background: High-dimensional proteomics data present significant challenges in biomarker discovery due to technical noise, feature redundancy, and multicollinearity. Current feature selection methods, including filter, wrapper, and embedded approaches, struggle with stability, sparsity, and computational efficiency. To address these limitations, we propose Soft-Thresholded Compressed Sensing (ST-CS), a hybrid framework integrating 1-bit compressed sensing with K-Medoids clustering. Unlike conventional methods relying on manual thresholds, ST-CS automates feature selection by dynamically partitioning coefficient magnitudes into discriminative biomarkers and noise.
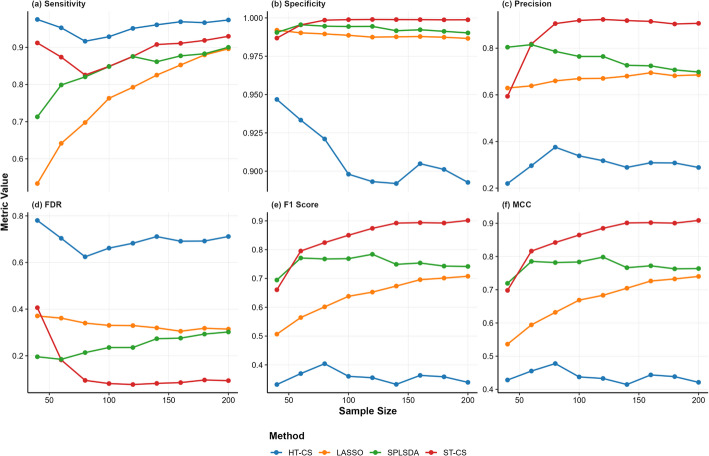
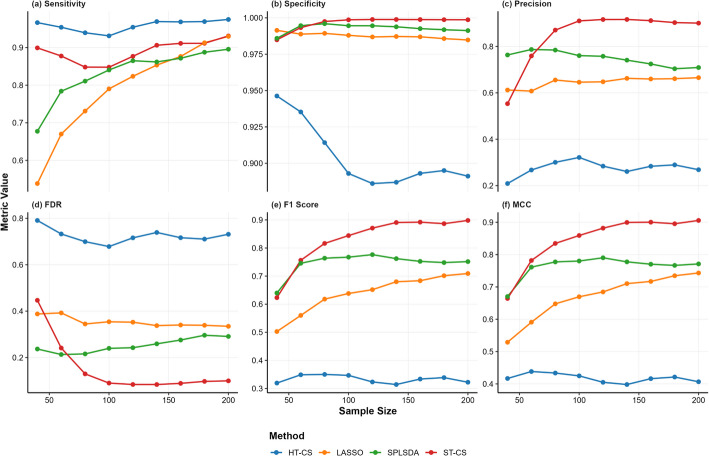
Results: Evaluations on simulated and real-world proteomic datasets demonstrated ST-CS's superiority in feature selection capability and classification performance. In simulations, ST-CS achieved feature selection robustness with balanced sensitivity (> 80%) and specificity (> 99.8%), reducing false discovery rates (FDR) by 20-50% compared to Hard-Thresholded Compressed Sensing (HT-CS). Additionally, it attained superior F1 scores and Matthews Correlation Coefficients (MCC), outperforming HT-CS, LASSO, and SPLSDA in identifying true biomarkers while suppressing noise. For classification performance, ST-CS surpassed all methods in the area under the receiver operating characteristic curve (AUC) across varying noise levels while maintaining sparsity. Applied to Clinical Proteomic Tumor Analysis Consortium (CPTAC) datasets, ST-CS matched HT-CS's classification accuracy (AUC = 97.47% for intrahepatic cholangiocarcinoma) but with 57% fewer selected features (37 vs. 86), demonstrating its dual strength in precision biomarker discovery and predictive accuracy. For glioblastoma data, ST-CS achieved higher AUC (72.71%) than HT-CS (72.15%), LASSO (67.80%), and SPLSDA (71.38%) while retaining a parsimonious feature set (30 vs. 58 features for HT-CS). In ovarian serous cystadenocarcinoma, ST-CS further demonstrated its adaptability, attaining superior AUC (75.86%) over HT-CS (75.61%), LASSO (61.00%), and SPLSDA (70.75%) with only 24 ± 5 selected biomarkers. These results highlight ST-CS's ability to rigorously automate feature selection while balancing classification efficacy, interpretability, and scalability for translational proteomics.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


