{"title":"使用混合控制实验对标和优化环境亚转录组学的定性和定量管道。","authors":"Weiyi Li, Qilian Fan, Yi Yang, Xiang Xiao, Jing Li, Yu Zhang","doi":"10.1093/ismeco/ycaf090","DOIUrl":null,"url":null,"abstract":"<p><p>Metatranscriptomic analysis is increasingly performed in environments to provide dynamic gene expression information on ecosystems, responding to their changing conditions. Many computational methods have undergone remarkable development in the past years, but a comprehensive benchmark study is still lacking. There are concerns regarding the accuracies of the qualitative and quantitative profilers obtained from metatranscriptomic analysis, especially for the microbiota in extreme environments, most of them are unculturable and lack well-annotated reference genomes. Here, we presented a benchmark experiment that included 10 single-species and their cell or RNA-admixtures with the predefined species compositions and varying evenness, simulating the low annotation rate and high heterogeneity. In total, 1 metagenome sample and 24 metatranscriptome were sequenced for the comparisons of 36 combination of analysis methods for tasks ranging from sample preparation, quality control, rRNA removal, alignment strategies, taxonomic profiling, and transcript quantification. For each part of the workflow mentioned above, corresponding metrics have been established to serve as standards for assessment and comparison. Evaluation revealed the performances and proposed an optimized pipeline named MT-Enviro (MetaTranscriptomic analysis for ENVIROnmental microbiome). Our data and analysis provide a comprehensive framework for benchmarking computational methods with metatranscriptomic analysis. MT-Enviro is implemented in Nextflow and is freely available from https://github.com/Li-Lab-SJTU/MT-Enviro.</p>","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":"5 1","pages":"ycaf090"},"PeriodicalIF":6.1000,"publicationDate":"2025-05-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12202999/pdf/","citationCount":"0","resultStr":"{\"title\":\"Benchmarking and optimizing qualitative and quantitative pipelines in environmental metatranscriptomics using mixture controlling experiments.\",\"authors\":\"Weiyi Li, Qilian Fan, Yi Yang, Xiang Xiao, Jing Li, Yu Zhang\",\"doi\":\"10.1093/ismeco/ycaf090\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Metatranscriptomic analysis is increasingly performed in environments to provide dynamic gene expression information on ecosystems, responding to their changing conditions. Many computational methods have undergone remarkable development in the past years, but a comprehensive benchmark study is still lacking. There are concerns regarding the accuracies of the qualitative and quantitative profilers obtained from metatranscriptomic analysis, especially for the microbiota in extreme environments, most of them are unculturable and lack well-annotated reference genomes. Here, we presented a benchmark experiment that included 10 single-species and their cell or RNA-admixtures with the predefined species compositions and varying evenness, simulating the low annotation rate and high heterogeneity. In total, 1 metagenome sample and 24 metatranscriptome were sequenced for the comparisons of 36 combination of analysis methods for tasks ranging from sample preparation, quality control, rRNA removal, alignment strategies, taxonomic profiling, and transcript quantification. For each part of the workflow mentioned above, corresponding metrics have been established to serve as standards for assessment and comparison. Evaluation revealed the performances and proposed an optimized pipeline named MT-Enviro (MetaTranscriptomic analysis for ENVIROnmental microbiome). Our data and analysis provide a comprehensive framework for benchmarking computational methods with metatranscriptomic analysis. MT-Enviro is implemented in Nextflow and is freely available from https://github.com/Li-Lab-SJTU/MT-Enviro.</p>\",\"PeriodicalId\":73516,\"journal\":{\"name\":\"ISME communications\",\"volume\":\"5 1\",\"pages\":\"ycaf090\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2025-05-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12202999/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISME communications\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/ismeco/ycaf090\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"ECOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/ismeco/ycaf090","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
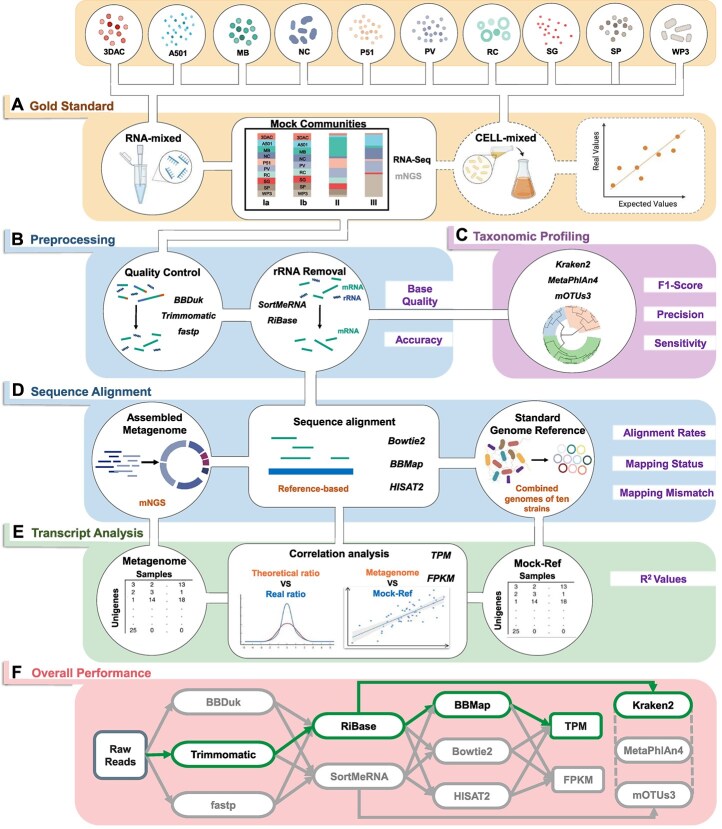
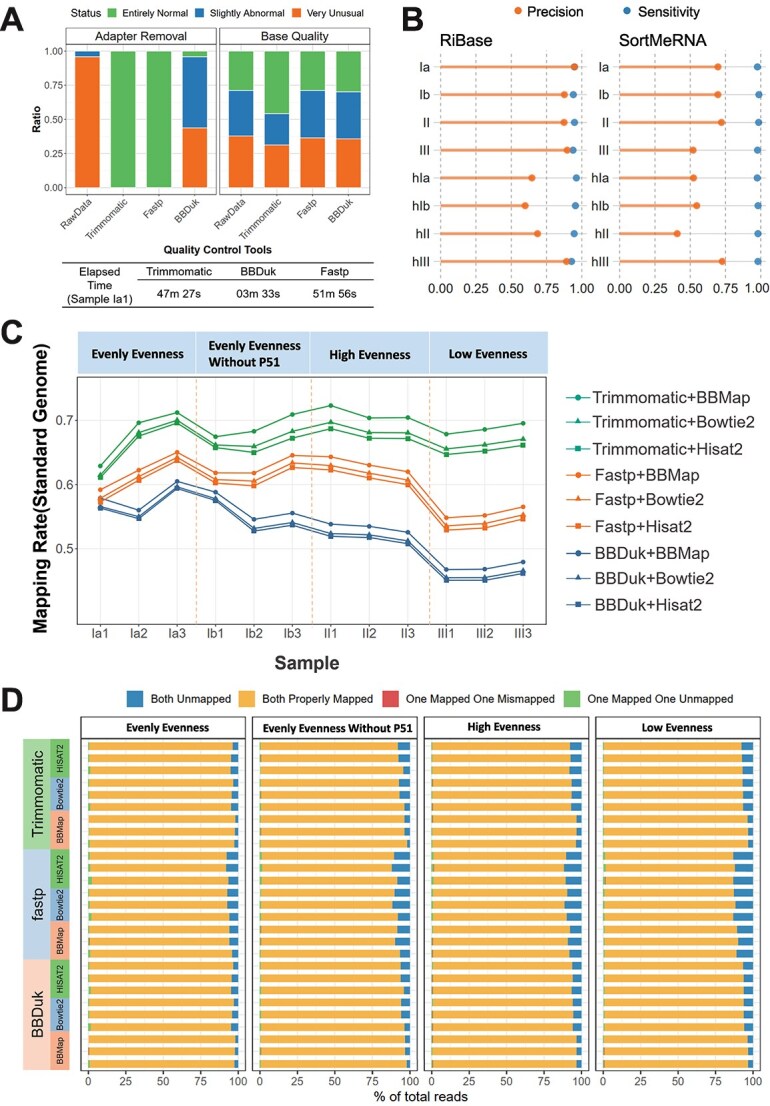
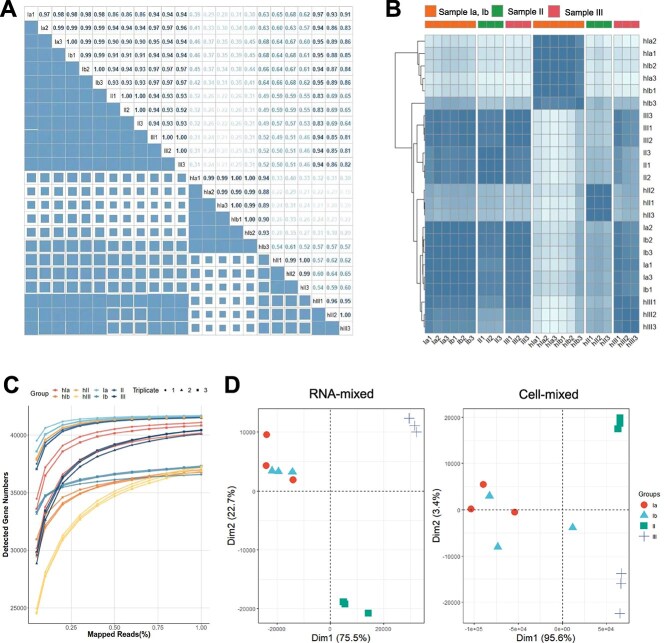
超转录组学分析越来越多地在环境中进行,以提供生态系统的动态基因表达信息,响应其变化的条件。近年来,许多计算方法取得了显著的发展,但仍缺乏全面的基准研究。人们对从超转录组学分析中获得的定性和定量谱图的准确性表示担忧,特别是对于极端环境中的微生物群,其中大多数是不可培养的,并且缺乏良好注释的参考基因组。在此,我们提出了一个基准实验,包括10个单一物种及其细胞或rna混合物,具有预定义的物种组成和不同的均匀度,模拟低注释率和高异质性。总共对1个宏基因组样本和24个亚转录组进行了测序,比较了36种分析方法的组合,包括样品制备、质量控制、rRNA去除、比对策略、分类分析和转录物定量。对于上面提到的工作流的每一部分,都建立了相应的度量,作为评估和比较的标准。评价结果显示了该方法的性能,并提出了一个优化的管道,命名为MT-Enviro (MetaTranscriptomic analysis for ENVIROnmental microbiome)。我们的数据和分析提供了一个全面的框架,为基准计算方法与亚转录组分析。MT-Enviro在Nextflow中实现,可以从https://github.com/Li-Lab-SJTU/MT-Enviro免费获得。
Benchmarking and optimizing qualitative and quantitative pipelines in environmental metatranscriptomics using mixture controlling experiments.
Metatranscriptomic analysis is increasingly performed in environments to provide dynamic gene expression information on ecosystems, responding to their changing conditions. Many computational methods have undergone remarkable development in the past years, but a comprehensive benchmark study is still lacking. There are concerns regarding the accuracies of the qualitative and quantitative profilers obtained from metatranscriptomic analysis, especially for the microbiota in extreme environments, most of them are unculturable and lack well-annotated reference genomes. Here, we presented a benchmark experiment that included 10 single-species and their cell or RNA-admixtures with the predefined species compositions and varying evenness, simulating the low annotation rate and high heterogeneity. In total, 1 metagenome sample and 24 metatranscriptome were sequenced for the comparisons of 36 combination of analysis methods for tasks ranging from sample preparation, quality control, rRNA removal, alignment strategies, taxonomic profiling, and transcript quantification. For each part of the workflow mentioned above, corresponding metrics have been established to serve as standards for assessment and comparison. Evaluation revealed the performances and proposed an optimized pipeline named MT-Enviro (MetaTranscriptomic analysis for ENVIROnmental microbiome). Our data and analysis provide a comprehensive framework for benchmarking computational methods with metatranscriptomic analysis. MT-Enviro is implemented in Nextflow and is freely available from https://github.com/Li-Lab-SJTU/MT-Enviro.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


