Rashid R. Valiev, Rinat T. Nasibullin, Hilda Sandström, Patrick Rinke, Kai Puolamäki and Theo Kurten
{"title":"基于结构描述符和机器学习的烷氧基自由基对系统间交叉速率常数预测","authors":"Rashid R. Valiev, Rinat T. Nasibullin, Hilda Sandström, Patrick Rinke, Kai Puolamäki and Theo Kurten","doi":"10.1039/D5CP01101A","DOIUrl":null,"url":null,"abstract":"<p >Peroxy radicals (RO<small><sub>2</sub></small>) are ubiquitous intermediates in many oxidation processes, especially in the atmospheric gas phase. The recombination reaction of two peroxy radicals (RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small>) has been demonstrated to lead, <em>via</em> several steps, to a triplet complex of two alkoxy radicals: <small><sup>3</sup></small>(RO˙⋯R′O˙). The different product channels of RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small> reactions thus correspond to different reactions of this triplet complex. Of particular interest to atmospheric chemistry is the intersystem crossing (ISC) to the singlet state, which enables the recombination of the two radicals to an ROOR′ peroxide with considerably lower volatility than the original precursors. These peroxides are believed to be key contributors to the formation of secondary organic aerosol (SOA) particles, which in turn contribute to both air pollution and radiative forcing uncertainties. Developing reliable computational models for, <em>e.g.</em>, RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small> branching ratios requires accurate estimates of the ISC rate constants, which can currently be obtained only from computationally expensive quantum chemistry calculations. By contrast, machine learning (ML) methods offer a faster alternative for estimating ISC rate constants. In the present work, we create a dataset with 98 082 conformations of radical pairs and their corresponding rate constants. We apply three ML models—random forest (RF), CatBoost (CB), and a neural network (NN)—to predict ISC rate constants from triplet to singlet states. Specifically, the models predict <em>k</em><small><sub>ISC</sub></small>(T<small><sub>1</sub></small> → S<small><sub>i</sub></small>) for <em>i</em> = 1–4 and the cumulative <em>k</em><small><sub>ISC</sub></small>(T<small><sub>1</sub></small> → S<small><sub><em>n</em></sub></small>), in alkoxy radical pairs, using only molecular geometry descriptors as inputs. All ML models achieved a mean absolute error (MAE) on our test set within one order of magnitude and a coefficient of determination <em>R</em><small><sup>2</sup></small> > 0.82 for all rate constants. Overall, the ML prediction matches the quantum chemical calculations within 1–2 orders of magnitude, providing a fast and scalable alternative for quantum chemical methods for ISC rate estimation.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 28","pages":" 14804-14814"},"PeriodicalIF":2.9000,"publicationDate":"2025-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp01101a?page=search","citationCount":"0","resultStr":"{\"title\":\"Predicting intersystem crossing rate constants of alkoxy-radical pairs with structure-based descriptors and machine learning†\",\"authors\":\"Rashid R. Valiev, Rinat T. Nasibullin, Hilda Sandström, Patrick Rinke, Kai Puolamäki and Theo Kurten\",\"doi\":\"10.1039/D5CP01101A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Peroxy radicals (RO<small><sub>2</sub></small>) are ubiquitous intermediates in many oxidation processes, especially in the atmospheric gas phase. The recombination reaction of two peroxy radicals (RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small>) has been demonstrated to lead, <em>via</em> several steps, to a triplet complex of two alkoxy radicals: <small><sup>3</sup></small>(RO˙⋯R′O˙). The different product channels of RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small> reactions thus correspond to different reactions of this triplet complex. Of particular interest to atmospheric chemistry is the intersystem crossing (ISC) to the singlet state, which enables the recombination of the two radicals to an ROOR′ peroxide with considerably lower volatility than the original precursors. These peroxides are believed to be key contributors to the formation of secondary organic aerosol (SOA) particles, which in turn contribute to both air pollution and radiative forcing uncertainties. Developing reliable computational models for, <em>e.g.</em>, RO<small><sub>2</sub></small> + R′O<small><sub>2</sub></small> branching ratios requires accurate estimates of the ISC rate constants, which can currently be obtained only from computationally expensive quantum chemistry calculations. By contrast, machine learning (ML) methods offer a faster alternative for estimating ISC rate constants. In the present work, we create a dataset with 98 082 conformations of radical pairs and their corresponding rate constants. We apply three ML models—random forest (RF), CatBoost (CB), and a neural network (NN)—to predict ISC rate constants from triplet to singlet states. Specifically, the models predict <em>k</em><small><sub>ISC</sub></small>(T<small><sub>1</sub></small> → S<small><sub>i</sub></small>) for <em>i</em> = 1–4 and the cumulative <em>k</em><small><sub>ISC</sub></small>(T<small><sub>1</sub></small> → S<small><sub><em>n</em></sub></small>), in alkoxy radical pairs, using only molecular geometry descriptors as inputs. All ML models achieved a mean absolute error (MAE) on our test set within one order of magnitude and a coefficient of determination <em>R</em><small><sup>2</sup></small> > 0.82 for all rate constants. Overall, the ML prediction matches the quantum chemical calculations within 1–2 orders of magnitude, providing a fast and scalable alternative for quantum chemical methods for ISC rate estimation.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 28\",\"pages\":\" 14804-14814\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp01101a?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01101a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01101a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
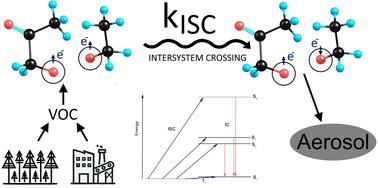
过氧化自由基(RO2)是许多氧化过程中普遍存在的中间体,特别是在大气气相中。两个过氧自由基(RO2 + R 'O2)的复合反应经过几个步骤,得到了两个烷氧自由基的三重络合物:3(RO●…R 'O●)。因此,RO2 + R 'O2反应的不同生成物通道对应于该三元络合物的不同反应。大气化学特别感兴趣的是系统间交叉(ISC)到单线态,这使得两个自由基重组为ROOR过氧化物,其挥发性比原始前体低得多。这些过氧化物被认为是二次有机气溶胶(SOA)颗粒形成的关键因素,而这反过来又导致了空气污染和辐射强迫的不确定性。开发可靠的计算模型,例如,RO2 + R 'O2分支比需要精确估计ISC速率常数,目前只能从计算昂贵的量子化学计算中获得。相比之下,机器学习(ML)方法为估计ISC速率常数提供了更快的替代方法。在目前的工作中,我们创建了一个包含98,082个自由基对构象及其相应的速率常数的数据集。我们应用三种机器学习模型——随机森林(RF)、CatBoost (CB)和神经网络(NN)来预测从三重态到单重态的ISC速率常数。具体而言,该模型仅使用分子几何描述符作为输入,预测了烷氧基自由基对中i = 1-4时的kISC(T1→Si)和累积kISC(T1→Sn)。所有ML模型在我们的测试集中实现了一个数量级的平均绝对误差(MAE)和决定系数R²>;所有速率常数为0.82。总体而言,ML预测与量子化学计算在1-2个数量级内匹配,为量子化学方法的ISC速率估计提供了一种快速且可扩展的替代方案。
Predicting intersystem crossing rate constants of alkoxy-radical pairs with structure-based descriptors and machine learning†
Peroxy radicals (RO2) are ubiquitous intermediates in many oxidation processes, especially in the atmospheric gas phase. The recombination reaction of two peroxy radicals (RO2 + R′O2) has been demonstrated to lead, via several steps, to a triplet complex of two alkoxy radicals: 3(RO˙⋯R′O˙). The different product channels of RO2 + R′O2 reactions thus correspond to different reactions of this triplet complex. Of particular interest to atmospheric chemistry is the intersystem crossing (ISC) to the singlet state, which enables the recombination of the two radicals to an ROOR′ peroxide with considerably lower volatility than the original precursors. These peroxides are believed to be key contributors to the formation of secondary organic aerosol (SOA) particles, which in turn contribute to both air pollution and radiative forcing uncertainties. Developing reliable computational models for, e.g., RO2 + R′O2 branching ratios requires accurate estimates of the ISC rate constants, which can currently be obtained only from computationally expensive quantum chemistry calculations. By contrast, machine learning (ML) methods offer a faster alternative for estimating ISC rate constants. In the present work, we create a dataset with 98 082 conformations of radical pairs and their corresponding rate constants. We apply three ML models—random forest (RF), CatBoost (CB), and a neural network (NN)—to predict ISC rate constants from triplet to singlet states. Specifically, the models predict kISC(T1 → Si) for i = 1–4 and the cumulative kISC(T1 → Sn), in alkoxy radical pairs, using only molecular geometry descriptors as inputs. All ML models achieved a mean absolute error (MAE) on our test set within one order of magnitude and a coefficient of determination R2 > 0.82 for all rate constants. Overall, the ML prediction matches the quantum chemical calculations within 1–2 orders of magnitude, providing a fast and scalable alternative for quantum chemical methods for ISC rate estimation.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


