Lian Duan, Kowit Hengphasatporn* and Yasuteru Shigeta,
{"title":"路径采样在MD和QM/MM MD仿真的Python工具包。","authors":"Lian Duan, Kowit Hengphasatporn* and Yasuteru Shigeta, ","doi":"10.1021/acs.jcim.5c00936","DOIUrl":null,"url":null,"abstract":"<p >PaCS-Q is an open-source Python toolkit that simplifies QM/MM MD and MD simulations, making complex pathway sampling accessible and user-friendly. Seamlessly integrated with the AMBER MD suite, it automates QM/MM MD simulations using the parallel cascade selection (PaCS) algorithm, enabling efficient exploration of reaction pathways without predefined reaction coordinates. PaCS-Q supports both RMSD- and distance-based sampling, which is ideal for studying covalent reactions and ligand binding/unbinding events. A key feature is its ability to automatically generate QM input files for Gaussian and ORCA directly from representative structures, streamlining the transition from MD to quantum calculations. With built-in tools for structure analysis and energy profiling, PaCS-Q minimizes setup complexity and enhances reproducibility. Easy to install via pip and compatible with Unix-based systems, PaCS-Q offers a practical, versatile solution for researchers in computational chemistry and drug discovery, enabling advanced simulations with speed, accuracy, and minimal effort. The PaCS-Q Python toolkit publicly available at https://github.com/nyelidl/PaCS-Q/.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 13","pages":"6441–6445"},"PeriodicalIF":5.3000,"publicationDate":"2025-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"PaCS-Q: Python Toolkits for Path Sampling in MD and QM/MM MD Simulation\",\"authors\":\"Lian Duan, Kowit Hengphasatporn* and Yasuteru Shigeta, \",\"doi\":\"10.1021/acs.jcim.5c00936\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >PaCS-Q is an open-source Python toolkit that simplifies QM/MM MD and MD simulations, making complex pathway sampling accessible and user-friendly. Seamlessly integrated with the AMBER MD suite, it automates QM/MM MD simulations using the parallel cascade selection (PaCS) algorithm, enabling efficient exploration of reaction pathways without predefined reaction coordinates. PaCS-Q supports both RMSD- and distance-based sampling, which is ideal for studying covalent reactions and ligand binding/unbinding events. A key feature is its ability to automatically generate QM input files for Gaussian and ORCA directly from representative structures, streamlining the transition from MD to quantum calculations. With built-in tools for structure analysis and energy profiling, PaCS-Q minimizes setup complexity and enhances reproducibility. Easy to install via pip and compatible with Unix-based systems, PaCS-Q offers a practical, versatile solution for researchers in computational chemistry and drug discovery, enabling advanced simulations with speed, accuracy, and minimal effort. The PaCS-Q Python toolkit publicly available at https://github.com/nyelidl/PaCS-Q/.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 13\",\"pages\":\"6441–6445\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-06-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00936\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00936","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
PaCS-Q: Python Toolkits for Path Sampling in MD and QM/MM MD Simulation
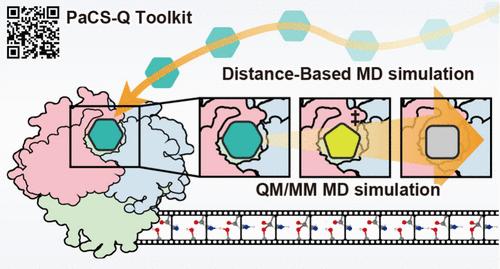
PaCS-Q is an open-source Python toolkit that simplifies QM/MM MD and MD simulations, making complex pathway sampling accessible and user-friendly. Seamlessly integrated with the AMBER MD suite, it automates QM/MM MD simulations using the parallel cascade selection (PaCS) algorithm, enabling efficient exploration of reaction pathways without predefined reaction coordinates. PaCS-Q supports both RMSD- and distance-based sampling, which is ideal for studying covalent reactions and ligand binding/unbinding events. A key feature is its ability to automatically generate QM input files for Gaussian and ORCA directly from representative structures, streamlining the transition from MD to quantum calculations. With built-in tools for structure analysis and energy profiling, PaCS-Q minimizes setup complexity and enhances reproducibility. Easy to install via pip and compatible with Unix-based systems, PaCS-Q offers a practical, versatile solution for researchers in computational chemistry and drug discovery, enabling advanced simulations with speed, accuracy, and minimal effort. The PaCS-Q Python toolkit publicly available at https://github.com/nyelidl/PaCS-Q/.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


