Mariya Levkova, Mihael Tsalta-Mladenov, Milena Stoyanova, Mari Hachmeriyan, Lyudmila Angelova, Ara Kaprelyan
{"title":"保加利亚瓦尔纳的亨廷顿舞蹈病二十年:临床趋势和挑战的回顾性单中心研究。","authors":"Mariya Levkova, Mihael Tsalta-Mladenov, Milena Stoyanova, Mari Hachmeriyan, Lyudmila Angelova, Ara Kaprelyan","doi":"10.3390/neurolint17060095","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background</b>: Huntington's disease (HD) is a progressive, autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the <i>HTT</i> gene. Despite advances in understanding its molecular basis, epidemiological data in many countries, including Bulgaria, remain limited. This study aims to present clinical and genetic findings from a 20-year single-centre cohort. <b>Methods</b>: A retrospective review was conducted of patients evaluated for HD at the University Hospital \"St. Marina\" in Varna between 2004 and 2024. Data included demographics, CAG repeat length, clinical features, imaging, and psychiatric assessments. Statistical analysis focused on correlations between variables, with significance set at <i>p</i> < 0.05. <b>Results</b>: Out of 79 referred individuals, 43 were molecularly confirmed. The mean age of onset was 43 years, with a four-year diagnostic delay. The average CAG repeat length was 44.6, though two symptomatic patients had reduced penetrance alleles (38 and 39 repeats). Cognitive and psychiatric symptoms were each present in 72% of cases. Depression was significantly more prevalent in women (<i>p</i> = 0.011). Most patients had a positive family history, predominantly maternal. <b>Conclusions</b>: Our findings highlight diagnostic delays, gender-specific psychiatric vulnerabilities, and the importance of personalized care. Improved access to genetic counselling and early diagnosis are essential for optimizing outcomes.</p>","PeriodicalId":19130,"journal":{"name":"Neurology International","volume":"17 6","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2025-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12195989/pdf/","citationCount":"0","resultStr":"{\"title\":\"Two Decades of Huntington's Disease in Varna, Bulgaria: A Retrospective Single-Centre Study of Clinical Trends and Challenges.\",\"authors\":\"Mariya Levkova, Mihael Tsalta-Mladenov, Milena Stoyanova, Mari Hachmeriyan, Lyudmila Angelova, Ara Kaprelyan\",\"doi\":\"10.3390/neurolint17060095\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><b>Background</b>: Huntington's disease (HD) is a progressive, autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the <i>HTT</i> gene. Despite advances in understanding its molecular basis, epidemiological data in many countries, including Bulgaria, remain limited. This study aims to present clinical and genetic findings from a 20-year single-centre cohort. <b>Methods</b>: A retrospective review was conducted of patients evaluated for HD at the University Hospital \\\"St. Marina\\\" in Varna between 2004 and 2024. Data included demographics, CAG repeat length, clinical features, imaging, and psychiatric assessments. Statistical analysis focused on correlations between variables, with significance set at <i>p</i> < 0.05. <b>Results</b>: Out of 79 referred individuals, 43 were molecularly confirmed. The mean age of onset was 43 years, with a four-year diagnostic delay. The average CAG repeat length was 44.6, though two symptomatic patients had reduced penetrance alleles (38 and 39 repeats). Cognitive and psychiatric symptoms were each present in 72% of cases. Depression was significantly more prevalent in women (<i>p</i> = 0.011). Most patients had a positive family history, predominantly maternal. <b>Conclusions</b>: Our findings highlight diagnostic delays, gender-specific psychiatric vulnerabilities, and the importance of personalized care. Improved access to genetic counselling and early diagnosis are essential for optimizing outcomes.</p>\",\"PeriodicalId\":19130,\"journal\":{\"name\":\"Neurology International\",\"volume\":\"17 6\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2025-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12195989/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology International\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/neurolint17060095\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology International","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/neurolint17060095","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Two Decades of Huntington's Disease in Varna, Bulgaria: A Retrospective Single-Centre Study of Clinical Trends and Challenges.
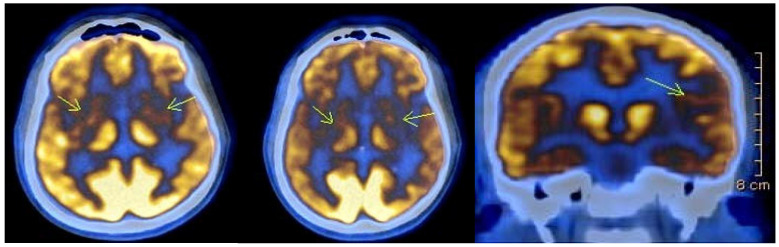
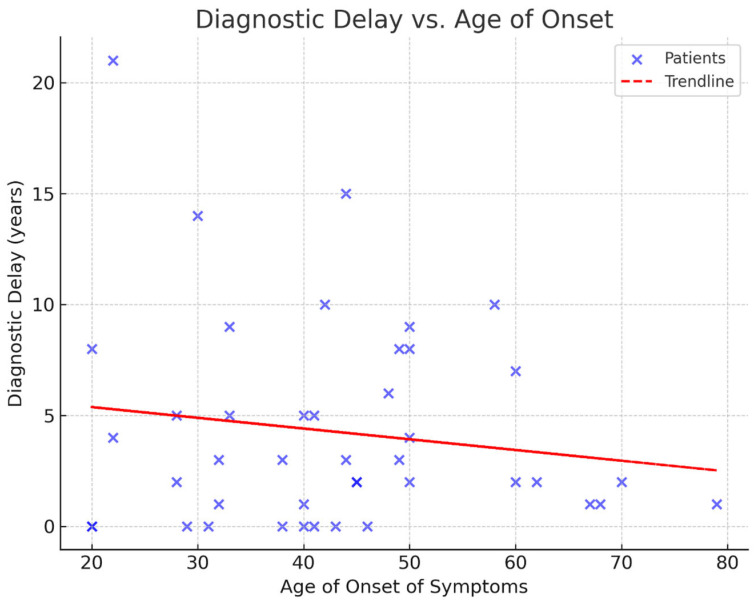
Background: Huntington's disease (HD) is a progressive, autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the HTT gene. Despite advances in understanding its molecular basis, epidemiological data in many countries, including Bulgaria, remain limited. This study aims to present clinical and genetic findings from a 20-year single-centre cohort. Methods: A retrospective review was conducted of patients evaluated for HD at the University Hospital "St. Marina" in Varna between 2004 and 2024. Data included demographics, CAG repeat length, clinical features, imaging, and psychiatric assessments. Statistical analysis focused on correlations between variables, with significance set at p < 0.05. Results: Out of 79 referred individuals, 43 were molecularly confirmed. The mean age of onset was 43 years, with a four-year diagnostic delay. The average CAG repeat length was 44.6, though two symptomatic patients had reduced penetrance alleles (38 and 39 repeats). Cognitive and psychiatric symptoms were each present in 72% of cases. Depression was significantly more prevalent in women (p = 0.011). Most patients had a positive family history, predominantly maternal. Conclusions: Our findings highlight diagnostic delays, gender-specific psychiatric vulnerabilities, and the importance of personalized care. Improved access to genetic counselling and early diagnosis are essential for optimizing outcomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


