{"title":"含五氮鎓阳离子盐的热化学性质评价。","authors":"D. V. Khakimov, A. A. Voronin","doi":"10.1007/s00894-025-06420-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Using quantum-chemical and crystal modeling, the solid-state enthalpies of three hypothetical high-energy salts with the pentazenium cation N<sub>5</sub><sup>+</sup> were estimated: nitrate NO<sub>3</sub><sup>−</sup>, dinitramide N(NO<sub>2</sub>)<sub>2</sub><sup>−</sup>, and azide N<sub>3</sub><sup>−</sup>, yielding the cationic contribution of pentazenium. Supplementing the lattice energy mixing method with an additive approach to determining the enthalpies of salts, values are given for salts with the perchlorate anion N<sub>5</sub><sup>+</sup>ClO<sub>4</sub><sup>−</sup>; four salts with the halogen anions N<sub>5</sub><sup>+</sup>I<sup>−</sup>, N<sub>5</sub><sup>+</sup>Br<sup>−</sup>, N<sub>5</sub><sup>+</sup>Cl<sup>−</sup>, and N<sub>5</sub><sup>+</sup>F<sup>−</sup>; and two experimentally existing structures: pentazenium tetrafluoroborate N<sub>5</sub><sup>+</sup>BF<sub>4</sub><sup>−</sup> and pentazenium hexafluorophosphate N<sub>5</sub><sup>+</sup>PF<sub>6</sub><sup>−</sup>. Calculations of salt structures were performed using a wide range of density functional theory (DFT) methods with varying functionals and bases, as well as composite methods for calculating the enthalpies of gas phase formation, to show the low variability of the final result and the universality of the approach. The calculations of the explosive characteristics of salts with oxygen–nitrogen anions showed that their detonation velocity is in the range of 7.1–7.6 km s<sup>−1</sup>. Of the three salts considered, the azide salt has the lowest density equal to 1.1 g cm<sup>−3</sup>, while the nitrate and dinitramide are at the level of 1.6 g cm<sup>−3</sup> and 1.7 g cm<sup>−3</sup>, respectively. The heat of detonation of pentazenium salts is about 800–850 cal g<sup>−1</sup>.</p><h3>Methods</h3><p>In this work, broad set of DFT calculations were conducted through the software Gaussian 09: B3LYP/6-31G(d,p), B3LYP/aug-cc-PVDZ + GD2, M052X/aug-cc-pVTZ, and M062X/6–311 + + G(d,p). For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP, and enthalpies of formation in gas phase were assessed by CBS-4 M, G3B3, G4, and W1BD.\n</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 7","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Evaluation of thermochemical characteristics of salts with pentazenium cation\",\"authors\":\"D. V. Khakimov, A. A. Voronin\",\"doi\":\"10.1007/s00894-025-06420-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Using quantum-chemical and crystal modeling, the solid-state enthalpies of three hypothetical high-energy salts with the pentazenium cation N<sub>5</sub><sup>+</sup> were estimated: nitrate NO<sub>3</sub><sup>−</sup>, dinitramide N(NO<sub>2</sub>)<sub>2</sub><sup>−</sup>, and azide N<sub>3</sub><sup>−</sup>, yielding the cationic contribution of pentazenium. Supplementing the lattice energy mixing method with an additive approach to determining the enthalpies of salts, values are given for salts with the perchlorate anion N<sub>5</sub><sup>+</sup>ClO<sub>4</sub><sup>−</sup>; four salts with the halogen anions N<sub>5</sub><sup>+</sup>I<sup>−</sup>, N<sub>5</sub><sup>+</sup>Br<sup>−</sup>, N<sub>5</sub><sup>+</sup>Cl<sup>−</sup>, and N<sub>5</sub><sup>+</sup>F<sup>−</sup>; and two experimentally existing structures: pentazenium tetrafluoroborate N<sub>5</sub><sup>+</sup>BF<sub>4</sub><sup>−</sup> and pentazenium hexafluorophosphate N<sub>5</sub><sup>+</sup>PF<sub>6</sub><sup>−</sup>. Calculations of salt structures were performed using a wide range of density functional theory (DFT) methods with varying functionals and bases, as well as composite methods for calculating the enthalpies of gas phase formation, to show the low variability of the final result and the universality of the approach. The calculations of the explosive characteristics of salts with oxygen–nitrogen anions showed that their detonation velocity is in the range of 7.1–7.6 km s<sup>−1</sup>. Of the three salts considered, the azide salt has the lowest density equal to 1.1 g cm<sup>−3</sup>, while the nitrate and dinitramide are at the level of 1.6 g cm<sup>−3</sup> and 1.7 g cm<sup>−3</sup>, respectively. The heat of detonation of pentazenium salts is about 800–850 cal g<sup>−1</sup>.</p><h3>Methods</h3><p>In this work, broad set of DFT calculations were conducted through the software Gaussian 09: B3LYP/6-31G(d,p), B3LYP/aug-cc-PVDZ + GD2, M052X/aug-cc-pVTZ, and M062X/6–311 + + G(d,p). For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP, and enthalpies of formation in gas phase were assessed by CBS-4 M, G3B3, G4, and W1BD.\\n</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 7\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-06-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06420-w\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06420-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:利用量子化学和晶体模型,估计了三种假设的高能盐的固态焓与五氮鎓阳离子N5+:硝酸盐NO3-,二硝酰胺N(NO2)2-和叠氮化物N3-,产生五氮鎓的阳离子贡献。在晶格能混合法的基础上,用加法法测定盐的焓值,给出了含有高氯酸盐阴离子N5+ClO4-的盐的焓值;四种盐与卤素阴离子N5+I-, N5+Br-, N5+Cl-和N5+F-;以及两种实验存在的结构:四氟硼酸五氮铵N5+BF4-和六氟磷酸五氮铵N5+PF6-。盐结构的计算使用了多种具有不同泛函和基的密度泛函理论(DFT)方法,以及计算气相形成焓的复合方法,以显示最终结果的低可变性和方法的普遍性。氧氮阴离子盐的爆炸特性计算表明,其爆速在7.1 ~ 7.6 km s-1范围内。在考虑的三种盐中,叠氮盐的密度最低,为1.1 g cm-3,而硝酸盐和二硝酰胺的密度分别为1.6 g cm-3和1.7 g cm-3。五氮盐的爆热约为800-850卡g-1。方法:通过Gaussian 09软件:B3LYP/6-31G(d,p)、B3LYP/ augc -cc- pvdz + GD2、M052X/ augc -cc- pvtz和M062X/6-311 + + G(d,p)进行广泛的DFT计算。在晶体结构优化方面,采用PMC (Packing of Molecules in crystal)程序的原子-原子势方法。用FitMEP拟合分子静电势电荷,用cbs - 4m、G3B3、G4和W1BD测定气相生成焓。
Evaluation of thermochemical characteristics of salts with pentazenium cation
Context
Using quantum-chemical and crystal modeling, the solid-state enthalpies of three hypothetical high-energy salts with the pentazenium cation N5+ were estimated: nitrate NO3−, dinitramide N(NO2)2−, and azide N3−, yielding the cationic contribution of pentazenium. Supplementing the lattice energy mixing method with an additive approach to determining the enthalpies of salts, values are given for salts with the perchlorate anion N5+ClO4−; four salts with the halogen anions N5+I−, N5+Br−, N5+Cl−, and N5+F−; and two experimentally existing structures: pentazenium tetrafluoroborate N5+BF4− and pentazenium hexafluorophosphate N5+PF6−. Calculations of salt structures were performed using a wide range of density functional theory (DFT) methods with varying functionals and bases, as well as composite methods for calculating the enthalpies of gas phase formation, to show the low variability of the final result and the universality of the approach. The calculations of the explosive characteristics of salts with oxygen–nitrogen anions showed that their detonation velocity is in the range of 7.1–7.6 km s−1. Of the three salts considered, the azide salt has the lowest density equal to 1.1 g cm−3, while the nitrate and dinitramide are at the level of 1.6 g cm−3 and 1.7 g cm−3, respectively. The heat of detonation of pentazenium salts is about 800–850 cal g−1.
Methods
In this work, broad set of DFT calculations were conducted through the software Gaussian 09: B3LYP/6-31G(d,p), B3LYP/aug-cc-PVDZ + GD2, M052X/aug-cc-pVTZ, and M062X/6–311 + + G(d,p). For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP, and enthalpies of formation in gas phase were assessed by CBS-4 M, G3B3, G4, and W1BD.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.
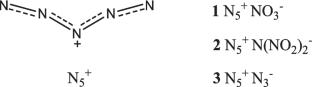

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


