Sergio Alfaro, Fabián González-Norambuena, José Luis Velázquez-Libera, Francisco Adasme-Carreño* and Julio Caballero*,
{"title":"CorrEA:一个考虑多种结合位点构象的配体-受体系统中计算能量和活性之间关系优化的Web服务器。","authors":"Sergio Alfaro, Fabián González-Norambuena, José Luis Velázquez-Libera, Francisco Adasme-Carreño* and Julio Caballero*, ","doi":"10.1021/acs.jcim.5c00571","DOIUrl":null,"url":null,"abstract":"<p ><i>In silico</i> molecular models of receptor–ligand complexes, built using molecular docking methods, are valuable as they potentially reveal the chemical interactions responsible for specific affinities. When applied to series of congeneric compounds, they help formulate theories about the effects of different substituents on affinity differences. Molecular docking provides (i) a pose where chemical interactions are optimized and (ii) an energy value indicating how favorable the interaction is. The capability of molecular docking for the first purpose is recognized, but it fails considerably in the second. It is widely known that energy values obtained by molecular docking are unreliable, which makes their application to congeneric series unable to correlate these computationally calculated energy values with laboratory-derived biological activities. Theoretically, an improved correlation could be obtained when protein flexibility is considered in the docking calculation; i.e., flexibility in the protein residues at the binding site can give access to more representative docking solutions. With this in mind, in this work, we present the novel web server CorrEA with a simple and innovative way of considering the flexibility of ligand–protein systems. To apply the method, users must generate a set of receptor conformations exhibiting significant variability within the binding site. Subsequently, they should cross-dock the ligand series they intend to study, to obtain various poses for each ligand across the different receptor conformations. CorrEA performs a genetic algorithm (GA) selection to extract a representative complex for each ligand that better adjusts the global correlation between calculated docking energy values and experimental logarithmic biological activities. In the end, CorrEA provides the ligand–protein pairs that produce the highest correlations. The new method was tested in several different cases to demonstrate its usefulness.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 13","pages":"7113–7128"},"PeriodicalIF":5.3000,"publicationDate":"2025-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"CorrEA: A Web Server for Optimizing Correlations between Calculated Energies and Activities in Ligand–Receptor Systems Considering Multiple Binding Site Conformations\",\"authors\":\"Sergio Alfaro, Fabián González-Norambuena, José Luis Velázquez-Libera, Francisco Adasme-Carreño* and Julio Caballero*, \",\"doi\":\"10.1021/acs.jcim.5c00571\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p ><i>In silico</i> molecular models of receptor–ligand complexes, built using molecular docking methods, are valuable as they potentially reveal the chemical interactions responsible for specific affinities. When applied to series of congeneric compounds, they help formulate theories about the effects of different substituents on affinity differences. Molecular docking provides (i) a pose where chemical interactions are optimized and (ii) an energy value indicating how favorable the interaction is. The capability of molecular docking for the first purpose is recognized, but it fails considerably in the second. It is widely known that energy values obtained by molecular docking are unreliable, which makes their application to congeneric series unable to correlate these computationally calculated energy values with laboratory-derived biological activities. Theoretically, an improved correlation could be obtained when protein flexibility is considered in the docking calculation; i.e., flexibility in the protein residues at the binding site can give access to more representative docking solutions. With this in mind, in this work, we present the novel web server CorrEA with a simple and innovative way of considering the flexibility of ligand–protein systems. To apply the method, users must generate a set of receptor conformations exhibiting significant variability within the binding site. Subsequently, they should cross-dock the ligand series they intend to study, to obtain various poses for each ligand across the different receptor conformations. CorrEA performs a genetic algorithm (GA) selection to extract a representative complex for each ligand that better adjusts the global correlation between calculated docking energy values and experimental logarithmic biological activities. In the end, CorrEA provides the ligand–protein pairs that produce the highest correlations. The new method was tested in several different cases to demonstrate its usefulness.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 13\",\"pages\":\"7113–7128\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-06-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00571\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00571","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
CorrEA: A Web Server for Optimizing Correlations between Calculated Energies and Activities in Ligand–Receptor Systems Considering Multiple Binding Site Conformations
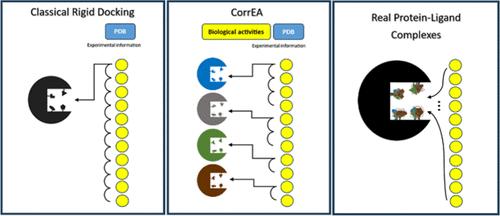
In silico molecular models of receptor–ligand complexes, built using molecular docking methods, are valuable as they potentially reveal the chemical interactions responsible for specific affinities. When applied to series of congeneric compounds, they help formulate theories about the effects of different substituents on affinity differences. Molecular docking provides (i) a pose where chemical interactions are optimized and (ii) an energy value indicating how favorable the interaction is. The capability of molecular docking for the first purpose is recognized, but it fails considerably in the second. It is widely known that energy values obtained by molecular docking are unreliable, which makes their application to congeneric series unable to correlate these computationally calculated energy values with laboratory-derived biological activities. Theoretically, an improved correlation could be obtained when protein flexibility is considered in the docking calculation; i.e., flexibility in the protein residues at the binding site can give access to more representative docking solutions. With this in mind, in this work, we present the novel web server CorrEA with a simple and innovative way of considering the flexibility of ligand–protein systems. To apply the method, users must generate a set of receptor conformations exhibiting significant variability within the binding site. Subsequently, they should cross-dock the ligand series they intend to study, to obtain various poses for each ligand across the different receptor conformations. CorrEA performs a genetic algorithm (GA) selection to extract a representative complex for each ligand that better adjusts the global correlation between calculated docking energy values and experimental logarithmic biological activities. In the end, CorrEA provides the ligand–protein pairs that produce the highest correlations. The new method was tested in several different cases to demonstrate its usefulness.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


