Hengwei Bian, Xueguang Shao, Christophe Chipot, Wensheng Cai, Haohao Fu
{"title":"一种高通量绝对自由束缚能计算的正式精确方法。","authors":"Hengwei Bian, Xueguang Shao, Christophe Chipot, Wensheng Cai, Haohao Fu","doi":"10.1038/s43588-025-00821-w","DOIUrl":null,"url":null,"abstract":"Here we introduce a high-throughput, formally exact method for absolute binding-free-energy calculations that enhances computational efficiency and accuracy. At the core of this method is a thermodynamic cycle that minimizes protein ligand relative motion, thereby reducing system perturbations and driving a fourfold gain in efficiency over the traditional double-decoupling method. By combining this strategy with double-wide sampling and hydrogen-mass repartitioning algorithms, the efficiency is further boosted to eightfold. The presented method is applied to 45 diverse protein–ligand complexes. For 34 complexes with validated force-field accuracy, our method achieves an average unsigned error of less than 1 kcal mol−1 and a hysteresis below 0.5 kcal mol−1, showcasing exceptional reliability. Moreover, it efficiently manages flexible peptide ligands through a potential-of-mean-force calculation, adding less than 5% extra simulation time. For 11 challenging cases, the presented method also shows an improvement compared with previously published results. Put together, this method has potential for advancing research in physical, biological and medicinal chemistry. A thermodynamic cycle is proposed that limits protein–ligand relative motion and boosts the efficiency of absolute binding-free-energy calculations four- to eightfold, without compromising theoretical rigor.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"5 8","pages":"621-626"},"PeriodicalIF":18.3000,"publicationDate":"2025-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A formally exact method for high-throughput absolute binding-free-energy calculations\",\"authors\":\"Hengwei Bian, Xueguang Shao, Christophe Chipot, Wensheng Cai, Haohao Fu\",\"doi\":\"10.1038/s43588-025-00821-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Here we introduce a high-throughput, formally exact method for absolute binding-free-energy calculations that enhances computational efficiency and accuracy. At the core of this method is a thermodynamic cycle that minimizes protein ligand relative motion, thereby reducing system perturbations and driving a fourfold gain in efficiency over the traditional double-decoupling method. By combining this strategy with double-wide sampling and hydrogen-mass repartitioning algorithms, the efficiency is further boosted to eightfold. The presented method is applied to 45 diverse protein–ligand complexes. For 34 complexes with validated force-field accuracy, our method achieves an average unsigned error of less than 1 kcal mol−1 and a hysteresis below 0.5 kcal mol−1, showcasing exceptional reliability. Moreover, it efficiently manages flexible peptide ligands through a potential-of-mean-force calculation, adding less than 5% extra simulation time. For 11 challenging cases, the presented method also shows an improvement compared with previously published results. Put together, this method has potential for advancing research in physical, biological and medicinal chemistry. A thermodynamic cycle is proposed that limits protein–ligand relative motion and boosts the efficiency of absolute binding-free-energy calculations four- to eightfold, without compromising theoretical rigor.\",\"PeriodicalId\":74246,\"journal\":{\"name\":\"Nature computational science\",\"volume\":\"5 8\",\"pages\":\"621-626\"},\"PeriodicalIF\":18.3000,\"publicationDate\":\"2025-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature computational science\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43588-025-00821-w\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-025-00821-w","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
A formally exact method for high-throughput absolute binding-free-energy calculations
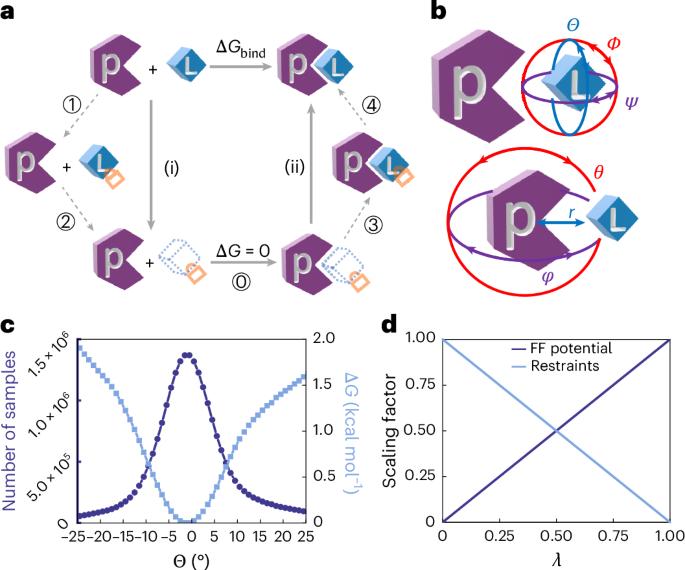
Here we introduce a high-throughput, formally exact method for absolute binding-free-energy calculations that enhances computational efficiency and accuracy. At the core of this method is a thermodynamic cycle that minimizes protein ligand relative motion, thereby reducing system perturbations and driving a fourfold gain in efficiency over the traditional double-decoupling method. By combining this strategy with double-wide sampling and hydrogen-mass repartitioning algorithms, the efficiency is further boosted to eightfold. The presented method is applied to 45 diverse protein–ligand complexes. For 34 complexes with validated force-field accuracy, our method achieves an average unsigned error of less than 1 kcal mol−1 and a hysteresis below 0.5 kcal mol−1, showcasing exceptional reliability. Moreover, it efficiently manages flexible peptide ligands through a potential-of-mean-force calculation, adding less than 5% extra simulation time. For 11 challenging cases, the presented method also shows an improvement compared with previously published results. Put together, this method has potential for advancing research in physical, biological and medicinal chemistry. A thermodynamic cycle is proposed that limits protein–ligand relative motion and boosts the efficiency of absolute binding-free-energy calculations four- to eightfold, without compromising theoretical rigor.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


