Amin Khodaei, Sania Eskandari, Hadi Sharifi, Behzad Mozaffari-Tazehkand
{"title":"PRCFX-DT:一种新的基于图的基因组序列特征选择和分类方法。","authors":"Amin Khodaei, Sania Eskandari, Hadi Sharifi, Behzad Mozaffari-Tazehkand","doi":"10.1186/s12859-025-06183-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In recent years, viral diseases have exhibited a significant incidence of infections and fatalities. The analysis of viral genomic sequences can be efficacious in evaluating the present and potentially forthcoming condition of viruses. Considering the importance of the internal structure of the cell and the nucleotide sequences within it, analyzing nucleotide sequences can provide a range of discussable features. On the other hand, it has been demonstrated that the use of graph algorithms and machine learning in the analysis and examination of virus samples and even viral variants can yield beneficial results.</p><p><strong>Results: </strong>This study proposes a novel approach that utilizes complex networks and probabilistic graph modeling methods to analyze viral genomic sequences for feature extraction. The proposed approach, which relies on the PageRank centrality algorithm, operates on codons that are associated with the nucleotide sequences. Experiments with machine learning algorithms were conducted on multiple datasets of viruses and various variants of coronavirus and influenza viruses. The use of a decision tree classifier model on the extracted distinguishing features enabled the differentiation of coronavirus samples from other samples. The high discriminative capability of the graph node centrality feature played a significant role in these experiments, establishing a meaningful connection with genetic concepts as well. The decision tree classifier applied on 173,228 genomic sequence samples originating from 30 distinct virus types, showed a remarkable accuracy rate of 99.73%.</p><p><strong>Conclusion: </strong>The proposed algorithm was successfully tested on several types of viruses, and the interpretability of the extracted features also enabled its structural analysis. The use of a graph-based approach on genetic features containing information about the internal structure of nucleotides yielded results that could be significant for the identification of any type of virus or specific viral variant.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"159"},"PeriodicalIF":3.3000,"publicationDate":"2025-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12172359/pdf/","citationCount":"0","resultStr":"{\"title\":\"PRCFX-DT: a new graph-based approach for feature selection and classification of genomic sequences.\",\"authors\":\"Amin Khodaei, Sania Eskandari, Hadi Sharifi, Behzad Mozaffari-Tazehkand\",\"doi\":\"10.1186/s12859-025-06183-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>In recent years, viral diseases have exhibited a significant incidence of infections and fatalities. The analysis of viral genomic sequences can be efficacious in evaluating the present and potentially forthcoming condition of viruses. Considering the importance of the internal structure of the cell and the nucleotide sequences within it, analyzing nucleotide sequences can provide a range of discussable features. On the other hand, it has been demonstrated that the use of graph algorithms and machine learning in the analysis and examination of virus samples and even viral variants can yield beneficial results.</p><p><strong>Results: </strong>This study proposes a novel approach that utilizes complex networks and probabilistic graph modeling methods to analyze viral genomic sequences for feature extraction. The proposed approach, which relies on the PageRank centrality algorithm, operates on codons that are associated with the nucleotide sequences. Experiments with machine learning algorithms were conducted on multiple datasets of viruses and various variants of coronavirus and influenza viruses. The use of a decision tree classifier model on the extracted distinguishing features enabled the differentiation of coronavirus samples from other samples. The high discriminative capability of the graph node centrality feature played a significant role in these experiments, establishing a meaningful connection with genetic concepts as well. The decision tree classifier applied on 173,228 genomic sequence samples originating from 30 distinct virus types, showed a remarkable accuracy rate of 99.73%.</p><p><strong>Conclusion: </strong>The proposed algorithm was successfully tested on several types of viruses, and the interpretability of the extracted features also enabled its structural analysis. The use of a graph-based approach on genetic features containing information about the internal structure of nucleotides yielded results that could be significant for the identification of any type of virus or specific viral variant.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"159\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-06-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12172359/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06183-4\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06183-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
PRCFX-DT: a new graph-based approach for feature selection and classification of genomic sequences.
Background: In recent years, viral diseases have exhibited a significant incidence of infections and fatalities. The analysis of viral genomic sequences can be efficacious in evaluating the present and potentially forthcoming condition of viruses. Considering the importance of the internal structure of the cell and the nucleotide sequences within it, analyzing nucleotide sequences can provide a range of discussable features. On the other hand, it has been demonstrated that the use of graph algorithms and machine learning in the analysis and examination of virus samples and even viral variants can yield beneficial results.
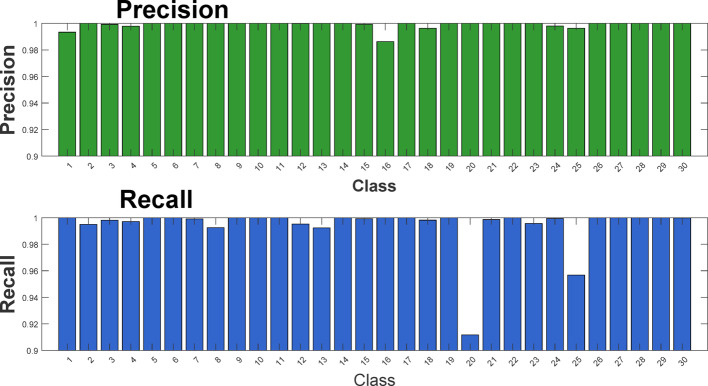
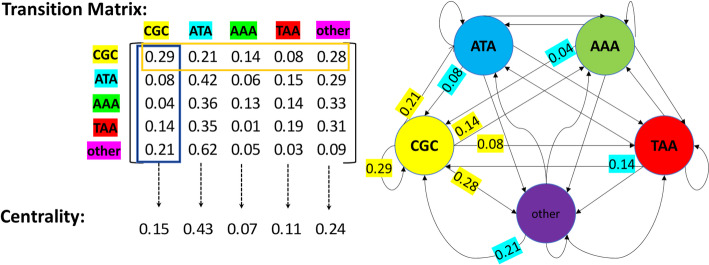
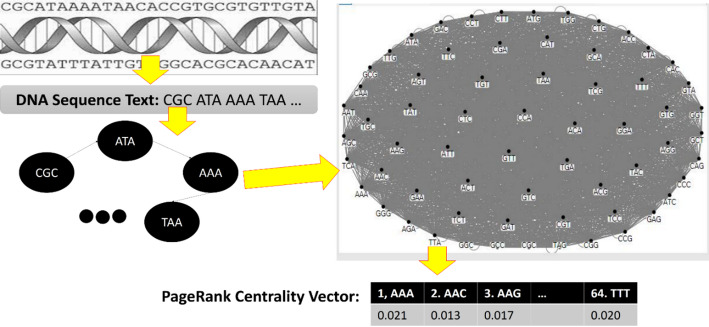
Results: This study proposes a novel approach that utilizes complex networks and probabilistic graph modeling methods to analyze viral genomic sequences for feature extraction. The proposed approach, which relies on the PageRank centrality algorithm, operates on codons that are associated with the nucleotide sequences. Experiments with machine learning algorithms were conducted on multiple datasets of viruses and various variants of coronavirus and influenza viruses. The use of a decision tree classifier model on the extracted distinguishing features enabled the differentiation of coronavirus samples from other samples. The high discriminative capability of the graph node centrality feature played a significant role in these experiments, establishing a meaningful connection with genetic concepts as well. The decision tree classifier applied on 173,228 genomic sequence samples originating from 30 distinct virus types, showed a remarkable accuracy rate of 99.73%.
Conclusion: The proposed algorithm was successfully tested on several types of viruses, and the interpretability of the extracted features also enabled its structural analysis. The use of a graph-based approach on genetic features containing information about the internal structure of nucleotides yielded results that could be significant for the identification of any type of virus or specific viral variant.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


