{"title":"探索Mo2C在锂氮电池中的催化作用","authors":"Lixin Xiong, and , Neil Qiang Su*, ","doi":"10.1021/acs.jpcc.5c0111010.1021/acs.jpcc.5c01110","DOIUrl":null,"url":null,"abstract":"<p >The rechargeable lithium–nitrogen (Li–N<sub>2</sub>) battery represents a promising strategy for nitrogen fixation and next-generation energy storage systems. However, the strong N≡N triple bond and high ionization energy pose significant challenges for an efficient nitrogen reduction reaction. This study employs density functional theory calculations to evaluate the performance of the Mo<sub>2</sub>C monolayer as a cathode catalyst for the Li–N<sub>2</sub> battery. Results reveal that N<sub>2</sub> interacts strongly with the Mo<sub>2</sub>C surface, significantly activating the N≡N bond and reducing the dissociation energy barrier to 0.379 eV. Crucially, Mo<sub>2</sub>C exhibits weak and strong adsorption of Li and N atoms, respectively, ensuring that active sites preferentially bond with N without Li poisoning. The calculated low charge/discharge overpotential (<0.62 V) indicates that Mo<sub>2</sub>C effectively facilitates N<sub>2</sub> evolution and reduction reactions. According to the ab initio molecular dynamics simulation, N<sub>2</sub> dissociation and Li<sub>3</sub>N formation can be successfully observed on Mo<sub>2</sub>C but not on graphene (frequently utilized in Li–O<sub>2</sub> and Li–CO<sub>2</sub> batteries). This study identifies the primary role of cathode catalysts as promoting the discharge reaction and reversible cycling in the Li–N<sub>2</sub> battery, rather than merely reducing overpotential. These findings provide new insights into catalyst design and optimization strategies for enhancing the performance of Li–N<sub>2</sub> batteries.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 24","pages":"10886–10899 10886–10899"},"PeriodicalIF":3.2000,"publicationDate":"2025-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring the Catalytic Role of Mo2C in Lithium–Nitrogen Batteries\",\"authors\":\"Lixin Xiong, and , Neil Qiang Su*, \",\"doi\":\"10.1021/acs.jpcc.5c0111010.1021/acs.jpcc.5c01110\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The rechargeable lithium–nitrogen (Li–N<sub>2</sub>) battery represents a promising strategy for nitrogen fixation and next-generation energy storage systems. However, the strong N≡N triple bond and high ionization energy pose significant challenges for an efficient nitrogen reduction reaction. This study employs density functional theory calculations to evaluate the performance of the Mo<sub>2</sub>C monolayer as a cathode catalyst for the Li–N<sub>2</sub> battery. Results reveal that N<sub>2</sub> interacts strongly with the Mo<sub>2</sub>C surface, significantly activating the N≡N bond and reducing the dissociation energy barrier to 0.379 eV. Crucially, Mo<sub>2</sub>C exhibits weak and strong adsorption of Li and N atoms, respectively, ensuring that active sites preferentially bond with N without Li poisoning. The calculated low charge/discharge overpotential (<0.62 V) indicates that Mo<sub>2</sub>C effectively facilitates N<sub>2</sub> evolution and reduction reactions. According to the ab initio molecular dynamics simulation, N<sub>2</sub> dissociation and Li<sub>3</sub>N formation can be successfully observed on Mo<sub>2</sub>C but not on graphene (frequently utilized in Li–O<sub>2</sub> and Li–CO<sub>2</sub> batteries). This study identifies the primary role of cathode catalysts as promoting the discharge reaction and reversible cycling in the Li–N<sub>2</sub> battery, rather than merely reducing overpotential. These findings provide new insights into catalyst design and optimization strategies for enhancing the performance of Li–N<sub>2</sub> batteries.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 24\",\"pages\":\"10886–10899 10886–10899\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-06-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c01110\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c01110","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Exploring the Catalytic Role of Mo2C in Lithium–Nitrogen Batteries
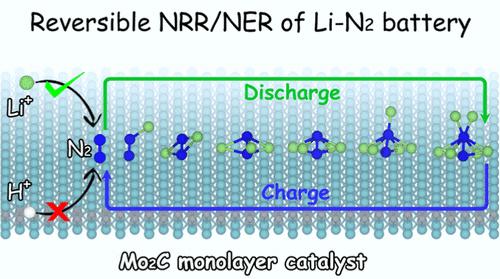
The rechargeable lithium–nitrogen (Li–N2) battery represents a promising strategy for nitrogen fixation and next-generation energy storage systems. However, the strong N≡N triple bond and high ionization energy pose significant challenges for an efficient nitrogen reduction reaction. This study employs density functional theory calculations to evaluate the performance of the Mo2C monolayer as a cathode catalyst for the Li–N2 battery. Results reveal that N2 interacts strongly with the Mo2C surface, significantly activating the N≡N bond and reducing the dissociation energy barrier to 0.379 eV. Crucially, Mo2C exhibits weak and strong adsorption of Li and N atoms, respectively, ensuring that active sites preferentially bond with N without Li poisoning. The calculated low charge/discharge overpotential (<0.62 V) indicates that Mo2C effectively facilitates N2 evolution and reduction reactions. According to the ab initio molecular dynamics simulation, N2 dissociation and Li3N formation can be successfully observed on Mo2C but not on graphene (frequently utilized in Li–O2 and Li–CO2 batteries). This study identifies the primary role of cathode catalysts as promoting the discharge reaction and reversible cycling in the Li–N2 battery, rather than merely reducing overpotential. These findings provide new insights into catalyst design and optimization strategies for enhancing the performance of Li–N2 batteries.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


