Yasuko Kobari, Non Miyata, Jun Takayama, Naoya Saijo, Tomohisa Suzuki, Shigeo Kure, Atsuo Kikuchi, Gen Tamiya, Takumi Takizawa
{"title":"日本Lenz-Majewski综合征伴PTDSS1新变异1例","authors":"Yasuko Kobari, Non Miyata, Jun Takayama, Naoya Saijo, Tomohisa Suzuki, Shigeo Kure, Atsuo Kikuchi, Gen Tamiya, Takumi Takizawa","doi":"10.1002/mgg3.70112","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lenz-Majewski syndrome (LMS) is a rare genetic disorder characterized by osteosclerosis, intellectual disability, characteristic facies, and distinct craniofacial, dental, cutaneous, and distal-limb anomalies. Mutations in the PTDSS1 gene, which encodes one of the phosphatidylserines (PS) synthase enzymes, PSS1, have been identified as causative in LMS patients. These mutations make PSS1 insensitive to feedback inhibition by PS levels.</p><p><strong>Methods: </strong>Whole genome sequence (WGS) was performed on a patient with congenital cutis laxa and her parents. PS synthase activity was analyzed in PTDSS1 mutant cDNA clones to evaluate functional alterations.</p><p><strong>Results: </strong>A 5-year-old girl presented with congenital skin wrinkles and was initially diagnosed with congenital cutis laxa. She had bilateral inner ear hypoplasia, bilateral low-frequency hearing loss, attention-deficit/hyperactivity disorder, and mild intellectual disability. Physical examination revealed protruding ears, frontal bossing, and dental malalignment. A de novo heterozygous missense variant in the PTDSS1 gene, c.284G>A (p. Arg95Gln) was identified by WGS. Functional analysis indicated increased PS synthase activity, supporting the pathogenicity of this variant.</p><p><strong>Conclusions: </strong>The patient's cutis laxa and facial features were consistent with LMS, though radiographic findings did not reveal the characteristic sclerosing bone dysplasia reported in previous cases. This observation suggests that LMS may have a broader phenotypic spectrum than previously recognized.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 6","pages":"e70112"},"PeriodicalIF":1.6000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12171241/pdf/","citationCount":"0","resultStr":"{\"title\":\"A Japanese Case of Lenz-Majewski Syndrome With a Novel PTDSS1 Variant.\",\"authors\":\"Yasuko Kobari, Non Miyata, Jun Takayama, Naoya Saijo, Tomohisa Suzuki, Shigeo Kure, Atsuo Kikuchi, Gen Tamiya, Takumi Takizawa\",\"doi\":\"10.1002/mgg3.70112\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lenz-Majewski syndrome (LMS) is a rare genetic disorder characterized by osteosclerosis, intellectual disability, characteristic facies, and distinct craniofacial, dental, cutaneous, and distal-limb anomalies. Mutations in the PTDSS1 gene, which encodes one of the phosphatidylserines (PS) synthase enzymes, PSS1, have been identified as causative in LMS patients. These mutations make PSS1 insensitive to feedback inhibition by PS levels.</p><p><strong>Methods: </strong>Whole genome sequence (WGS) was performed on a patient with congenital cutis laxa and her parents. PS synthase activity was analyzed in PTDSS1 mutant cDNA clones to evaluate functional alterations.</p><p><strong>Results: </strong>A 5-year-old girl presented with congenital skin wrinkles and was initially diagnosed with congenital cutis laxa. She had bilateral inner ear hypoplasia, bilateral low-frequency hearing loss, attention-deficit/hyperactivity disorder, and mild intellectual disability. Physical examination revealed protruding ears, frontal bossing, and dental malalignment. A de novo heterozygous missense variant in the PTDSS1 gene, c.284G>A (p. Arg95Gln) was identified by WGS. Functional analysis indicated increased PS synthase activity, supporting the pathogenicity of this variant.</p><p><strong>Conclusions: </strong>The patient's cutis laxa and facial features were consistent with LMS, though radiographic findings did not reveal the characteristic sclerosing bone dysplasia reported in previous cases. This observation suggests that LMS may have a broader phenotypic spectrum than previously recognized.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 6\",\"pages\":\"e70112\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12171241/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70112\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70112","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A Japanese Case of Lenz-Majewski Syndrome With a Novel PTDSS1 Variant.
Background: Lenz-Majewski syndrome (LMS) is a rare genetic disorder characterized by osteosclerosis, intellectual disability, characteristic facies, and distinct craniofacial, dental, cutaneous, and distal-limb anomalies. Mutations in the PTDSS1 gene, which encodes one of the phosphatidylserines (PS) synthase enzymes, PSS1, have been identified as causative in LMS patients. These mutations make PSS1 insensitive to feedback inhibition by PS levels.
Methods: Whole genome sequence (WGS) was performed on a patient with congenital cutis laxa and her parents. PS synthase activity was analyzed in PTDSS1 mutant cDNA clones to evaluate functional alterations.
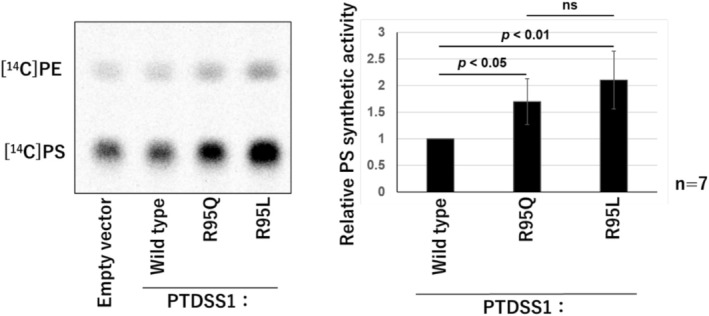
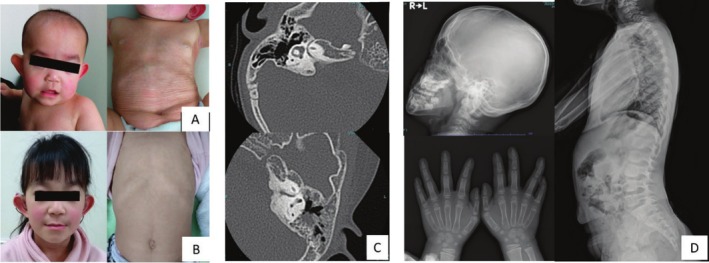
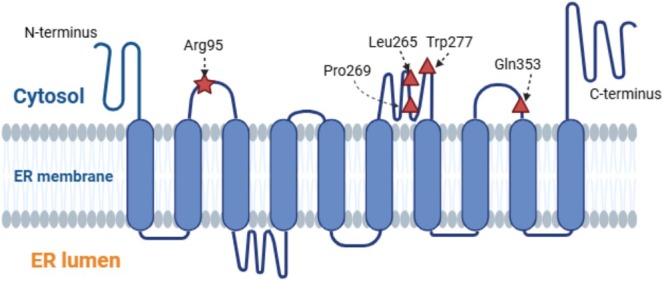
Results: A 5-year-old girl presented with congenital skin wrinkles and was initially diagnosed with congenital cutis laxa. She had bilateral inner ear hypoplasia, bilateral low-frequency hearing loss, attention-deficit/hyperactivity disorder, and mild intellectual disability. Physical examination revealed protruding ears, frontal bossing, and dental malalignment. A de novo heterozygous missense variant in the PTDSS1 gene, c.284G>A (p. Arg95Gln) was identified by WGS. Functional analysis indicated increased PS synthase activity, supporting the pathogenicity of this variant.
Conclusions: The patient's cutis laxa and facial features were consistent with LMS, though radiographic findings did not reveal the characteristic sclerosing bone dysplasia reported in previous cases. This observation suggests that LMS may have a broader phenotypic spectrum than previously recognized.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


