Hsiao-Jung Kao, Elin H F Wang, Erh-Chan Yeh, Hsiao-Huei Chen, Feng-Jen Hsieh, Tsang-Ming Ko, Wuh-Liang Hwu, Pui-Yan Kwok, Ni-Chung Lee
{"title":"通过全基因组分析鉴定Cornelia De Lange综合征患者nippl的新生染色体易位。","authors":"Hsiao-Jung Kao, Elin H F Wang, Erh-Chan Yeh, Hsiao-Huei Chen, Feng-Jen Hsieh, Tsang-Ming Ko, Wuh-Liang Hwu, Pui-Yan Kwok, Ni-Chung Lee","doi":"10.1002/mgg3.70115","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cornelia de Lange syndrome (CdLS) is a rare genetic disorder characterized by congenital multiple anomalies, developmental delay, and distinctive facial features.</p><p><strong>Methods: </strong>We performed chromosomal microarray analysis (CMA), whole exome sequencing (WES), linked-read whole-genome sequencing (WGS) and optical genome mapping (OGM) to investigate an undiagnosed case of CdLS.</p><p><strong>Results: </strong>A male patient presented clinical features consistent with CdLS, including a short nose, synophrys, small hands, hearing impairment, refractory complex partial seizures, and developmental delay. Amniocentesis at 28 gestational weeks and karyotyping revealed a presumably balanced translocation between chromosome 5 and chromosome 6. CMA and WES failed to identify copy number variants or a molecular diagnosis. Further analysis using WGS and OGM identified two translocation events on chromosome 5, resulting in three derivative chromosomes: 46,XY, der(2)t(2;5)(q32.3;p13.2),der(5)t(5;6)(p13.1;q12),der(6)t(2;6)(q32.3;q12)ins(6;5)(q12;p13.1p13.2). These rearrangements disrupted the NIPBL gene, a key gene with CdLS, splitting it across derivative chromosomes 2 and 6. Phasing studies revealed that these translocations originated from the paternal lineage.</p><p><strong>Conclusions: </strong>This case highlights the intricate genetic underpinnings of CdLS in this patient and underscores the diagnostic value of high-resolution genomic analyses in elucidating complex chromosomal rearrangements.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 6","pages":"e70115"},"PeriodicalIF":1.6000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12171785/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification of De Novo Chromosomal Translocations Disrupting NIPBL in a Patient With Cornelia de Lange Syndrome by Full Genome Analysis.\",\"authors\":\"Hsiao-Jung Kao, Elin H F Wang, Erh-Chan Yeh, Hsiao-Huei Chen, Feng-Jen Hsieh, Tsang-Ming Ko, Wuh-Liang Hwu, Pui-Yan Kwok, Ni-Chung Lee\",\"doi\":\"10.1002/mgg3.70115\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Cornelia de Lange syndrome (CdLS) is a rare genetic disorder characterized by congenital multiple anomalies, developmental delay, and distinctive facial features.</p><p><strong>Methods: </strong>We performed chromosomal microarray analysis (CMA), whole exome sequencing (WES), linked-read whole-genome sequencing (WGS) and optical genome mapping (OGM) to investigate an undiagnosed case of CdLS.</p><p><strong>Results: </strong>A male patient presented clinical features consistent with CdLS, including a short nose, synophrys, small hands, hearing impairment, refractory complex partial seizures, and developmental delay. Amniocentesis at 28 gestational weeks and karyotyping revealed a presumably balanced translocation between chromosome 5 and chromosome 6. CMA and WES failed to identify copy number variants or a molecular diagnosis. Further analysis using WGS and OGM identified two translocation events on chromosome 5, resulting in three derivative chromosomes: 46,XY, der(2)t(2;5)(q32.3;p13.2),der(5)t(5;6)(p13.1;q12),der(6)t(2;6)(q32.3;q12)ins(6;5)(q12;p13.1p13.2). These rearrangements disrupted the NIPBL gene, a key gene with CdLS, splitting it across derivative chromosomes 2 and 6. Phasing studies revealed that these translocations originated from the paternal lineage.</p><p><strong>Conclusions: </strong>This case highlights the intricate genetic underpinnings of CdLS in this patient and underscores the diagnostic value of high-resolution genomic analyses in elucidating complex chromosomal rearrangements.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 6\",\"pages\":\"e70115\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12171785/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70115\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70115","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:Cornelia de Lange综合征(CdLS)是一种罕见的遗传性疾病,其特征是先天性多发性异常、发育迟缓和明显的面部特征。方法:采用染色体微阵列分析(CMA)、全外显子组测序(WES)、连锁阅读全基因组测序(WGS)和光学基因组定位(OGM)对1例未确诊的CdLS进行研究。结果:1例男性患者表现出与CdLS一致的临床特征,包括短鼻、拇趾、小手、听力障碍、难治性复杂部分性癫痫和发育迟缓。28孕周羊膜穿刺术和核型分析显示5号染色体和6号染色体之间可能存在平衡易位。CMA和WES未能识别拷贝数变异或分子诊断。利用WGS和OGM进一步分析,在5号染色体上发现了两个易位事件,导致三条衍生染色体:46,XY, der(2)t(2;5)(q32.3;p13.2),der(5)t(5;6)(p13.1;q12),der(6)t(2;6)(q32.3;q12)ins(6;5)(q12;p13.1p13.2)。这些重排破坏了CdLS的关键基因NIPBL基因,将其分裂到衍生染色体2和6上。分阶段研究表明,这些易位起源于父系血统。结论:该病例强调了该患者CdLS的复杂遗传基础,并强调了高分辨率基因组分析在阐明复杂染色体重排中的诊断价值。
Identification of De Novo Chromosomal Translocations Disrupting NIPBL in a Patient With Cornelia de Lange Syndrome by Full Genome Analysis.
Background: Cornelia de Lange syndrome (CdLS) is a rare genetic disorder characterized by congenital multiple anomalies, developmental delay, and distinctive facial features.
Methods: We performed chromosomal microarray analysis (CMA), whole exome sequencing (WES), linked-read whole-genome sequencing (WGS) and optical genome mapping (OGM) to investigate an undiagnosed case of CdLS.
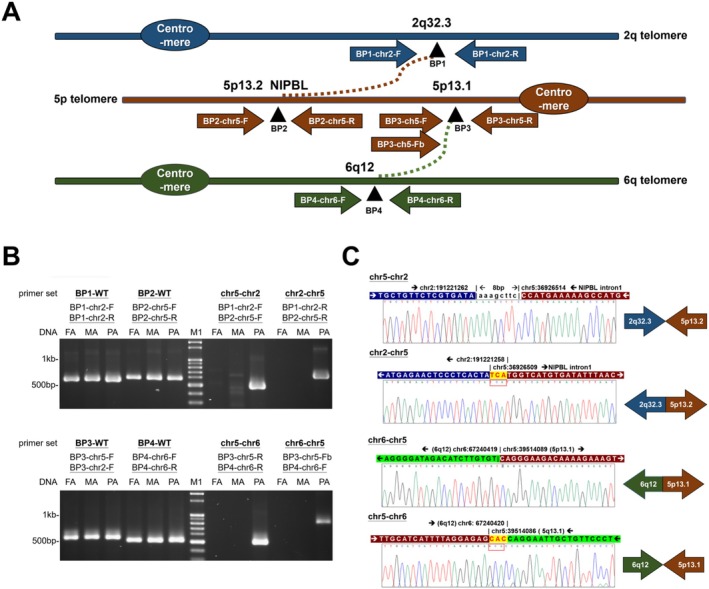
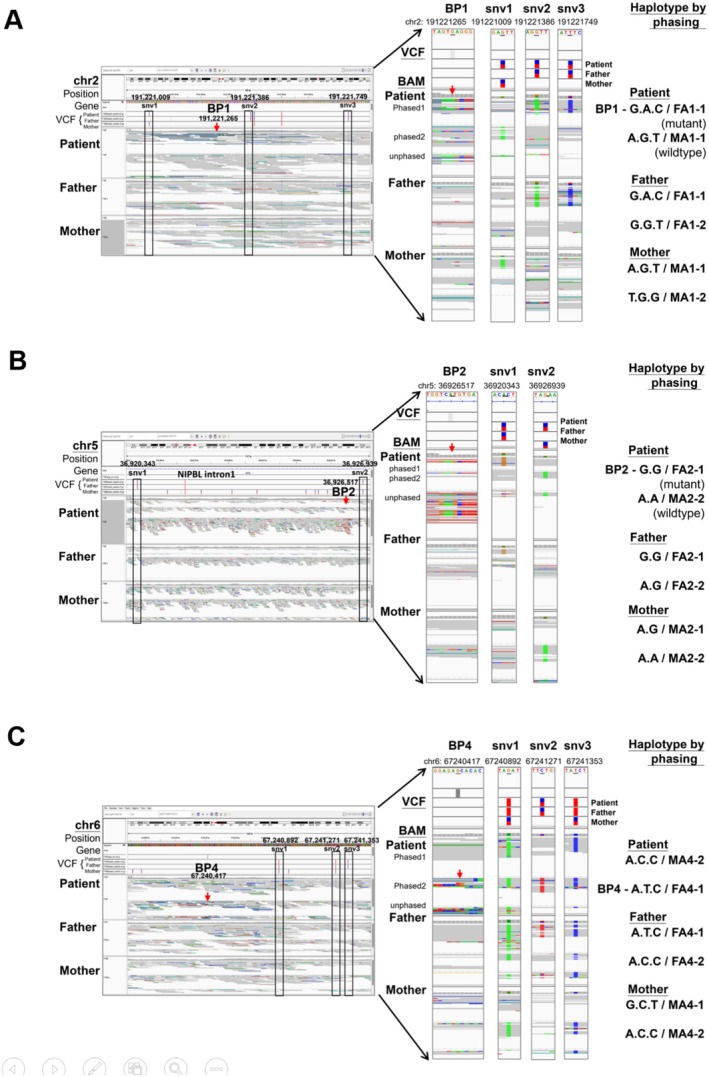
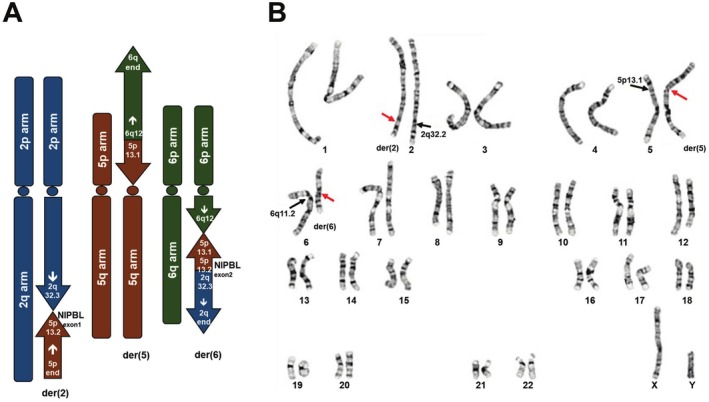
Results: A male patient presented clinical features consistent with CdLS, including a short nose, synophrys, small hands, hearing impairment, refractory complex partial seizures, and developmental delay. Amniocentesis at 28 gestational weeks and karyotyping revealed a presumably balanced translocation between chromosome 5 and chromosome 6. CMA and WES failed to identify copy number variants or a molecular diagnosis. Further analysis using WGS and OGM identified two translocation events on chromosome 5, resulting in three derivative chromosomes: 46,XY, der(2)t(2;5)(q32.3;p13.2),der(5)t(5;6)(p13.1;q12),der(6)t(2;6)(q32.3;q12)ins(6;5)(q12;p13.1p13.2). These rearrangements disrupted the NIPBL gene, a key gene with CdLS, splitting it across derivative chromosomes 2 and 6. Phasing studies revealed that these translocations originated from the paternal lineage.
Conclusions: This case highlights the intricate genetic underpinnings of CdLS in this patient and underscores the diagnostic value of high-resolution genomic analyses in elucidating complex chromosomal rearrangements.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


