{"title":"法洛四联症胎儿CCM2L中的复合杂合功能缺失变异。","authors":"Dandan Ling, Wanqin Xie, Xiao Mao, Zhiyu Liu, Yabing Tang, Fanjuan Kong","doi":"10.1002/mgg3.70117","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart disease. However, our current understanding of the genetic etiology for TOF is limited.</p><p><strong>Methods: </strong>Whole exome sequencing (WES) and Sanger sequencing were applied to a family trio diagnosed with TOF by fetal prenatal ultrasound examination. A minigene assay was performed to confirm the splicing defects.</p><p><strong>Results: </strong>We identified compound heterozygous variants in the cerebral cavernous malformation 2-like (CCM2L) gene, namely the paternally inherited nonsense variant NM_001365692.1:c.741G>A p.(Trp247Ter) and the maternally inherited splice-site variant NM_001365692.1:c.1263+2T>A in a fetus with TOF featuring a ventricular septal defect associated with overriding aorta and pulmonary stenosis. Minigene assay showed that the c.1263+2T>A variant led to skipping of CCM2L exon8 during RNA splicing, which is thought to result in frameshift and premature termination of translation. Both variants were absent from the public population databases (Genome Aggregation Database [gnomAD], 1000 Genomes [1000G], Clinvar) and classified as likely pathogenic according to the ACMG guidelines (PVS1 + PM2 level evidence).</p><p><strong>Conclusion: </strong>To our knowledge, this is the first reported case of biallelic loss-of-function variants in human CCM2L. Our findings suggest a potential association of human CCM2L with TOF.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 6","pages":"e70117"},"PeriodicalIF":1.6000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12168162/pdf/","citationCount":"0","resultStr":"{\"title\":\"Compound Heterozygous Loss-of-Function Variants in CCM2L in a Fetus With Tetralogy of Fallot.\",\"authors\":\"Dandan Ling, Wanqin Xie, Xiao Mao, Zhiyu Liu, Yabing Tang, Fanjuan Kong\",\"doi\":\"10.1002/mgg3.70117\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart disease. However, our current understanding of the genetic etiology for TOF is limited.</p><p><strong>Methods: </strong>Whole exome sequencing (WES) and Sanger sequencing were applied to a family trio diagnosed with TOF by fetal prenatal ultrasound examination. A minigene assay was performed to confirm the splicing defects.</p><p><strong>Results: </strong>We identified compound heterozygous variants in the cerebral cavernous malformation 2-like (CCM2L) gene, namely the paternally inherited nonsense variant NM_001365692.1:c.741G>A p.(Trp247Ter) and the maternally inherited splice-site variant NM_001365692.1:c.1263+2T>A in a fetus with TOF featuring a ventricular septal defect associated with overriding aorta and pulmonary stenosis. Minigene assay showed that the c.1263+2T>A variant led to skipping of CCM2L exon8 during RNA splicing, which is thought to result in frameshift and premature termination of translation. Both variants were absent from the public population databases (Genome Aggregation Database [gnomAD], 1000 Genomes [1000G], Clinvar) and classified as likely pathogenic according to the ACMG guidelines (PVS1 + PM2 level evidence).</p><p><strong>Conclusion: </strong>To our knowledge, this is the first reported case of biallelic loss-of-function variants in human CCM2L. Our findings suggest a potential association of human CCM2L with TOF.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 6\",\"pages\":\"e70117\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12168162/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70117\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70117","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Compound Heterozygous Loss-of-Function Variants in CCM2L in a Fetus With Tetralogy of Fallot.
Background: Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart disease. However, our current understanding of the genetic etiology for TOF is limited.
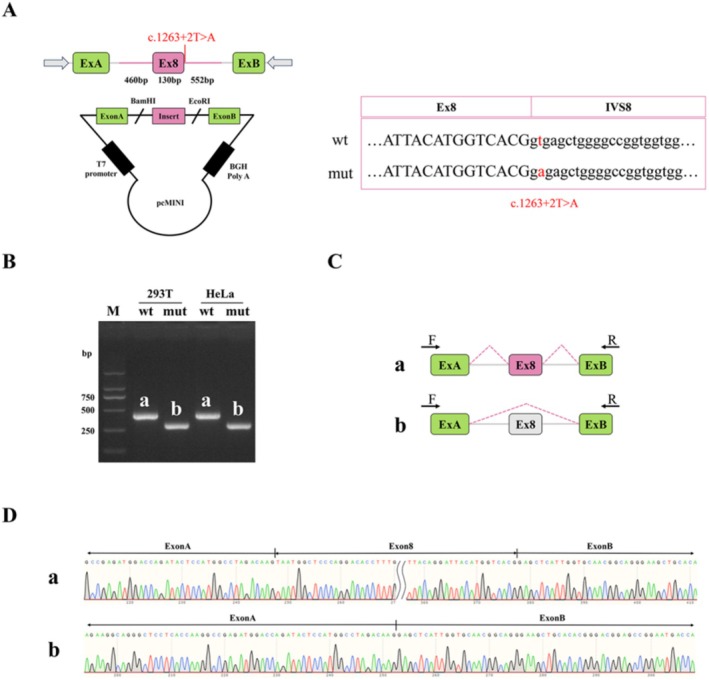
Methods: Whole exome sequencing (WES) and Sanger sequencing were applied to a family trio diagnosed with TOF by fetal prenatal ultrasound examination. A minigene assay was performed to confirm the splicing defects.
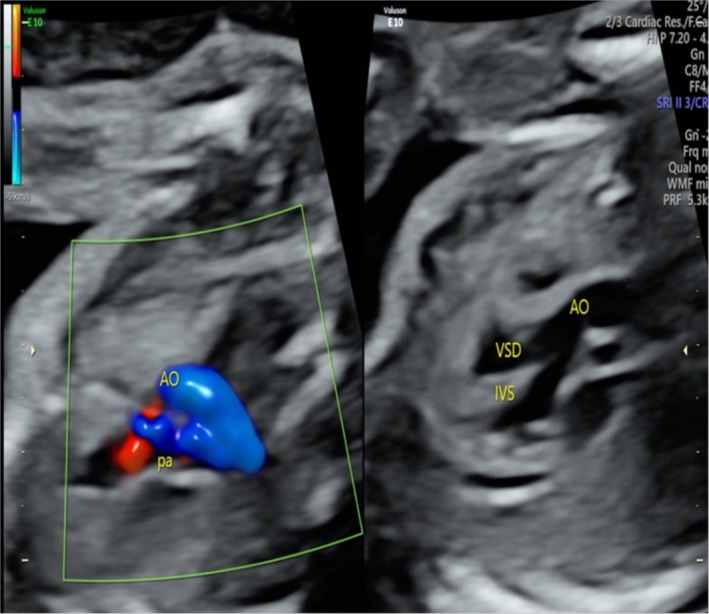
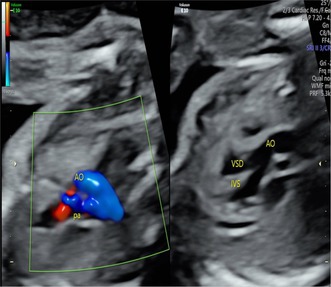
Results: We identified compound heterozygous variants in the cerebral cavernous malformation 2-like (CCM2L) gene, namely the paternally inherited nonsense variant NM_001365692.1:c.741G>A p.(Trp247Ter) and the maternally inherited splice-site variant NM_001365692.1:c.1263+2T>A in a fetus with TOF featuring a ventricular septal defect associated with overriding aorta and pulmonary stenosis. Minigene assay showed that the c.1263+2T>A variant led to skipping of CCM2L exon8 during RNA splicing, which is thought to result in frameshift and premature termination of translation. Both variants were absent from the public population databases (Genome Aggregation Database [gnomAD], 1000 Genomes [1000G], Clinvar) and classified as likely pathogenic according to the ACMG guidelines (PVS1 + PM2 level evidence).
Conclusion: To our knowledge, this is the first reported case of biallelic loss-of-function variants in human CCM2L. Our findings suggest a potential association of human CCM2L with TOF.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


