Rick Kamps, Herm Martens, Bart de Koning, Bert Smeets, Michel van Geel
{"title":"利用外显子组测序鉴定常染色体显性无染色体发育不全的一个新的致病FAM83H变异。","authors":"Rick Kamps, Herm Martens, Bart de Koning, Bert Smeets, Michel van Geel","doi":"10.1002/mgg3.70108","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Amelogenesis imperfecta (AI) is a rare genetic disorder causing tooth enamel defects. AI has been classified into 14 different clinical subtypes with different modes of inheritance. In this study, we performed whole-exome sequencing to identify the causative gene defect in a large Dutch family with autosomal dominant hypocalcified AI (ADHCAI).</p><p><strong>Methods: </strong>Whole-exome sequencing (WES) was performed on genomic DNA of the proband with AI. We focused on eight candidate genes known to be involved in inherited autosomal dominant AI. Sanger sequencing was used to confirm the selected exome candidate variant. Additionally, genotype and phenotype analyses were performed in the selected affected and non-affected individuals and compared according to previously listed literature for this candidate gene of the proband.</p><p><strong>Results: </strong>The clinical phenotype of the affected individuals showed a generalized and extensive enamel defect of all teeth. In the exome dataset of the proband, a novel nonsense variant in FAM83H, c.1055C>A p.(Ser352*) was detected, which was verified by conventional Sanger sequencing. Co-segregation analysis confirmed that the variant was present in all affected individuals and not in unaffected individuals.</p><p><strong>Conclusion: </strong>A novel pathogenic, protein-truncating variant was detected in FAM83H, a gene with similar truncating variants known to be associated with ADHCAI.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 6","pages":"e70108"},"PeriodicalIF":1.6000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12162354/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identifying a Novel Causal FAM83H Variant for Autosomal Dominant Amelogenesis Imperfecta Using Exome-Sequencing.\",\"authors\":\"Rick Kamps, Herm Martens, Bart de Koning, Bert Smeets, Michel van Geel\",\"doi\":\"10.1002/mgg3.70108\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Amelogenesis imperfecta (AI) is a rare genetic disorder causing tooth enamel defects. AI has been classified into 14 different clinical subtypes with different modes of inheritance. In this study, we performed whole-exome sequencing to identify the causative gene defect in a large Dutch family with autosomal dominant hypocalcified AI (ADHCAI).</p><p><strong>Methods: </strong>Whole-exome sequencing (WES) was performed on genomic DNA of the proband with AI. We focused on eight candidate genes known to be involved in inherited autosomal dominant AI. Sanger sequencing was used to confirm the selected exome candidate variant. Additionally, genotype and phenotype analyses were performed in the selected affected and non-affected individuals and compared according to previously listed literature for this candidate gene of the proband.</p><p><strong>Results: </strong>The clinical phenotype of the affected individuals showed a generalized and extensive enamel defect of all teeth. In the exome dataset of the proband, a novel nonsense variant in FAM83H, c.1055C>A p.(Ser352*) was detected, which was verified by conventional Sanger sequencing. Co-segregation analysis confirmed that the variant was present in all affected individuals and not in unaffected individuals.</p><p><strong>Conclusion: </strong>A novel pathogenic, protein-truncating variant was detected in FAM83H, a gene with similar truncating variants known to be associated with ADHCAI.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 6\",\"pages\":\"e70108\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12162354/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70108\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70108","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:无釉质发育不全症(Amelogenesis imperfecta, AI)是一种罕见的导致牙釉质缺陷的遗传性疾病。AI被分为14种不同的临床亚型,具有不同的遗传方式。在这项研究中,我们进行了全外显子组测序,以确定荷兰一个常染色体显性低钙化AI (ADHCAI)大家族的致病基因缺陷。方法:对AI先证者基因组DNA进行全外显子组测序(WES)。我们集中研究了八个已知与遗传性常染色体显性AI有关的候选基因。Sanger测序用于确认所选择的外显子组候选变异。此外,在选定的受影响和未受影响的个体中进行基因型和表型分析,并根据先前列出的先证者候选基因的文献进行比较。结果:患者临床表现为全身性、广泛性全牙釉质缺损。在先证的外显子组数据集中,检测到FAM83H, c.1055C> a p.(Ser352*)的一个新的无义变异,并通过常规Sanger测序进行验证。共分离分析证实该变异存在于所有受影响的个体中,而不存在于未受影响的个体中。结论:在FAM83H基因中检测到一种新的致病性蛋白截短变异,FAM83H基因具有与ADHCAI相关的类似截短变异。
Identifying a Novel Causal FAM83H Variant for Autosomal Dominant Amelogenesis Imperfecta Using Exome-Sequencing.
Background: Amelogenesis imperfecta (AI) is a rare genetic disorder causing tooth enamel defects. AI has been classified into 14 different clinical subtypes with different modes of inheritance. In this study, we performed whole-exome sequencing to identify the causative gene defect in a large Dutch family with autosomal dominant hypocalcified AI (ADHCAI).
Methods: Whole-exome sequencing (WES) was performed on genomic DNA of the proband with AI. We focused on eight candidate genes known to be involved in inherited autosomal dominant AI. Sanger sequencing was used to confirm the selected exome candidate variant. Additionally, genotype and phenotype analyses were performed in the selected affected and non-affected individuals and compared according to previously listed literature for this candidate gene of the proband.
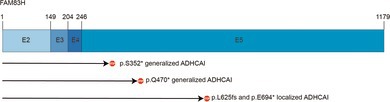
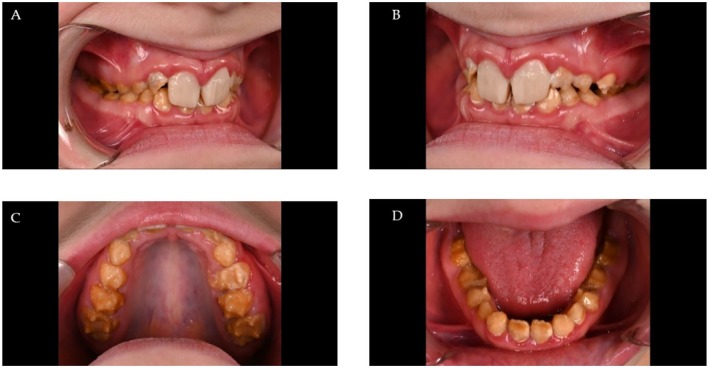
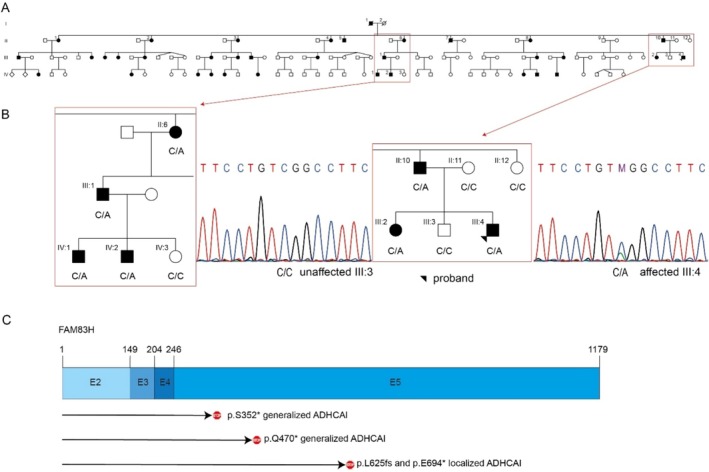
Results: The clinical phenotype of the affected individuals showed a generalized and extensive enamel defect of all teeth. In the exome dataset of the proband, a novel nonsense variant in FAM83H, c.1055C>A p.(Ser352*) was detected, which was verified by conventional Sanger sequencing. Co-segregation analysis confirmed that the variant was present in all affected individuals and not in unaffected individuals.
Conclusion: A novel pathogenic, protein-truncating variant was detected in FAM83H, a gene with similar truncating variants known to be associated with ADHCAI.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


