Natasha S Freeman, Kelsie Bogyo, Andrew Beenken, Jordan G Nestor, Simone Sanna-Cherchi, Pietro A Canetta
{"title":"家族性特发性肾小球疾病由myh9相关疾病的独特肾显性表型引起:一例报告。","authors":"Natasha S Freeman, Kelsie Bogyo, Andrew Beenken, Jordan G Nestor, Simone Sanna-Cherchi, Pietro A Canetta","doi":"10.1159/000546242","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>MYH9-related disease (MYH9-RD) is a rare genetic cause of proteinuric kidney disease. It typically manifests as a syndromic condition, presenting with macrocytic thrombocytopenia, sensorineural hearing loss (SNHL), chronic glomerulopathy, elevated liver enzymes, and early-onset bilateral cataracts. In accordance with CARE guidelines, we present a case report of a father and daughter with renal-predominant MYH9-RD due to a recently described missense variant affecting the head domain of non-muscle myosin heavy chain IIA.</p><p><strong>Case presentations: </strong>Patient 1 was an 18-year-old woman with childhood proteinuria who presented with severely advanced kidney failure. Dialysis was initiated as a bridge to a living unrelated renal transplant (LURT). Family history was notable for proteinuric kidney disease in her father (patient 2). Eight years later, genetic testing identified a likely pathogenic missense variant in the head domain of the <i>MYH9</i> gene (c.1271G>A, p.R424Q). A predictive structural model was obtained via AlphaFold Protein Structure Database, in which the mutation interrupts hydrogen bonding and π-cation interactions, likely leading to protein misfolding. Subsequent clinical screening revealed persistent mild thrombocytopenia and elevated liver enzymes, without cataracts or SNHL. Patient 2 was a 53-year-old man with childhood proteinuria who eventually presented with stage 4 chronic kidney disease and soon after underwent LURT. After patient 1's genetic diagnosis, he was confirmed to have the same mutation by genetic testing. Subsequent screening revealed mild thrombocytopenia and elevated liver enzymes with hepatic steatosis progressing to cirrhosis, without cataracts or SNHL.</p><p><strong>Conclusion: </strong>The finding of this <i>MYH9</i> p.R424Q variant confirmed a diagnosis of MYH9-RD in these patients. <i>MYH9</i> variants affecting the head domain typically result in severe thrombocytopenia. This recently reported head domain variant caused severe renal manifestations with mild thrombocytopenia and no manifestations of SNHL or cataracts in both patients, suggesting that this variant causes a renal-predominant form of MYH9-RD.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"5 1","pages":"243-249"},"PeriodicalIF":0.0000,"publicationDate":"2025-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12158415/pdf/","citationCount":"0","resultStr":"{\"title\":\"Familial Idiopathic Glomerular Disease due to a Unique Renal-Predominant Phenotype of MYH9-Related Disease: A Case Report.\",\"authors\":\"Natasha S Freeman, Kelsie Bogyo, Andrew Beenken, Jordan G Nestor, Simone Sanna-Cherchi, Pietro A Canetta\",\"doi\":\"10.1159/000546242\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>MYH9-related disease (MYH9-RD) is a rare genetic cause of proteinuric kidney disease. It typically manifests as a syndromic condition, presenting with macrocytic thrombocytopenia, sensorineural hearing loss (SNHL), chronic glomerulopathy, elevated liver enzymes, and early-onset bilateral cataracts. In accordance with CARE guidelines, we present a case report of a father and daughter with renal-predominant MYH9-RD due to a recently described missense variant affecting the head domain of non-muscle myosin heavy chain IIA.</p><p><strong>Case presentations: </strong>Patient 1 was an 18-year-old woman with childhood proteinuria who presented with severely advanced kidney failure. Dialysis was initiated as a bridge to a living unrelated renal transplant (LURT). Family history was notable for proteinuric kidney disease in her father (patient 2). Eight years later, genetic testing identified a likely pathogenic missense variant in the head domain of the <i>MYH9</i> gene (c.1271G>A, p.R424Q). A predictive structural model was obtained via AlphaFold Protein Structure Database, in which the mutation interrupts hydrogen bonding and π-cation interactions, likely leading to protein misfolding. Subsequent clinical screening revealed persistent mild thrombocytopenia and elevated liver enzymes, without cataracts or SNHL. Patient 2 was a 53-year-old man with childhood proteinuria who eventually presented with stage 4 chronic kidney disease and soon after underwent LURT. After patient 1's genetic diagnosis, he was confirmed to have the same mutation by genetic testing. Subsequent screening revealed mild thrombocytopenia and elevated liver enzymes with hepatic steatosis progressing to cirrhosis, without cataracts or SNHL.</p><p><strong>Conclusion: </strong>The finding of this <i>MYH9</i> p.R424Q variant confirmed a diagnosis of MYH9-RD in these patients. <i>MYH9</i> variants affecting the head domain typically result in severe thrombocytopenia. This recently reported head domain variant caused severe renal manifestations with mild thrombocytopenia and no manifestations of SNHL or cataracts in both patients, suggesting that this variant causes a renal-predominant form of MYH9-RD.</p>\",\"PeriodicalId\":73177,\"journal\":{\"name\":\"Glomerular diseases\",\"volume\":\"5 1\",\"pages\":\"243-249\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-05-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12158415/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Glomerular diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000546242\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000546242","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
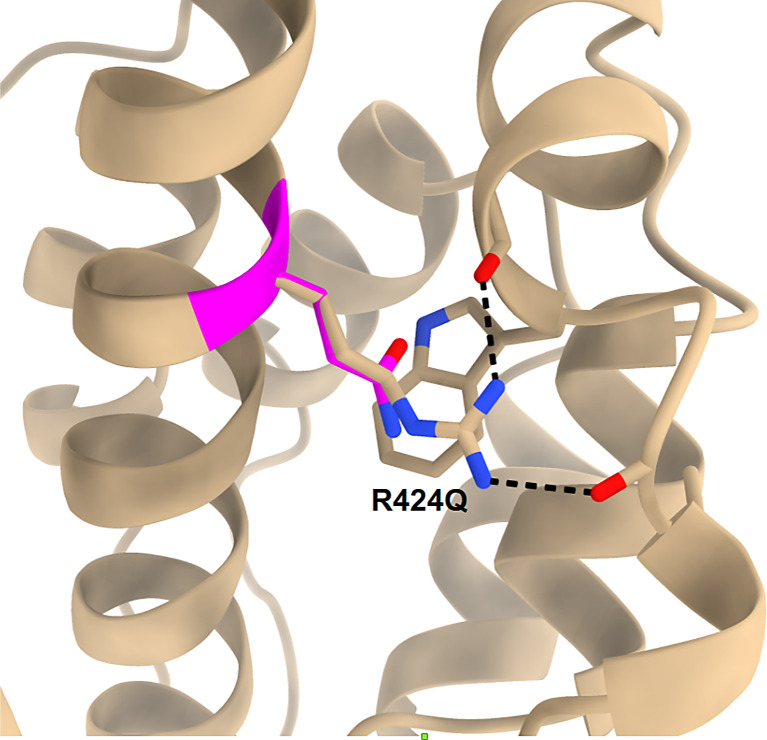
myh9相关疾病(MYH9-RD)是一种罕见的遗传性蛋白尿肾病。它通常表现为综合征,表现为大细胞血小板减少症、感音神经性听力损失(SNHL)、慢性肾小球病、肝酶升高和早发性双侧白内障。根据CARE指南,我们报告了一对父女因最近描述的影响非肌肉肌球蛋白重链IIA头部结构域的错义变异而患有肾脏显性MYH9-RD的病例。病例介绍:患者1是一名患有儿童期蛋白尿的18岁女性,表现为严重晚期肾衰竭。透析最初是作为非亲属活体肾移植(LURT)的桥梁。父亲有明显的蛋白尿肾病家族史(患者2)。8年后,基因检测发现MYH9基因头部结构域可能存在致病性错义变异(c.1271G> a, p.R424Q)。通过AlphaFold蛋白结构数据库获得预测结构模型,该突变中断了氢键和π-阳离子相互作用,可能导致蛋白质错误折叠。随后的临床筛查显示持续轻度血小板减少和肝酶升高,无白内障或SNHL。患者2是一名患有儿童期蛋白尿的53岁男性,最终表现为4期慢性肾脏疾病,并在接受LURT治疗后不久。患者1经基因诊断后,经基因检测证实具有相同的突变。随后的筛查显示轻度血小板减少症和肝酶升高伴肝脂肪变性进展为肝硬化,无白内障或SNHL。结论:MYH9 p.R424Q变异的发现证实了这些患者MYH9- rd的诊断。影响头部结构域的MYH9变异通常导致严重的血小板减少症。最近报道的这一头部结构域变异在两名患者中引起了严重的肾脏表现,伴有轻度血小板减少,但没有SNHL或白内障的表现,这表明该变异引起了MYH9-RD的肾脏显性形式。
Familial Idiopathic Glomerular Disease due to a Unique Renal-Predominant Phenotype of MYH9-Related Disease: A Case Report.
Introduction: MYH9-related disease (MYH9-RD) is a rare genetic cause of proteinuric kidney disease. It typically manifests as a syndromic condition, presenting with macrocytic thrombocytopenia, sensorineural hearing loss (SNHL), chronic glomerulopathy, elevated liver enzymes, and early-onset bilateral cataracts. In accordance with CARE guidelines, we present a case report of a father and daughter with renal-predominant MYH9-RD due to a recently described missense variant affecting the head domain of non-muscle myosin heavy chain IIA.
Case presentations: Patient 1 was an 18-year-old woman with childhood proteinuria who presented with severely advanced kidney failure. Dialysis was initiated as a bridge to a living unrelated renal transplant (LURT). Family history was notable for proteinuric kidney disease in her father (patient 2). Eight years later, genetic testing identified a likely pathogenic missense variant in the head domain of the MYH9 gene (c.1271G>A, p.R424Q). A predictive structural model was obtained via AlphaFold Protein Structure Database, in which the mutation interrupts hydrogen bonding and π-cation interactions, likely leading to protein misfolding. Subsequent clinical screening revealed persistent mild thrombocytopenia and elevated liver enzymes, without cataracts or SNHL. Patient 2 was a 53-year-old man with childhood proteinuria who eventually presented with stage 4 chronic kidney disease and soon after underwent LURT. After patient 1's genetic diagnosis, he was confirmed to have the same mutation by genetic testing. Subsequent screening revealed mild thrombocytopenia and elevated liver enzymes with hepatic steatosis progressing to cirrhosis, without cataracts or SNHL.
Conclusion: The finding of this MYH9 p.R424Q variant confirmed a diagnosis of MYH9-RD in these patients. MYH9 variants affecting the head domain typically result in severe thrombocytopenia. This recently reported head domain variant caused severe renal manifestations with mild thrombocytopenia and no manifestations of SNHL or cataracts in both patients, suggesting that this variant causes a renal-predominant form of MYH9-RD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


