Marie-Sophie C Ogloblinsky, Marc B Gros-La-Faige, Daniel P Lewinsohn, Mathilde Nguyen, Lourdes Velo-Suarez, Anthony Herzig, Thomas E Ludwig, Helen Castillo-Madeen, Donald F Conrad, Emmanuelle Génin, Gaëlle Marenne
{"title":"Easy-PSAP:一个集成的工作流程,优先考虑来自单个个体的序列数据中的致病变异。","authors":"Marie-Sophie C Ogloblinsky, Marc B Gros-La-Faige, Daniel P Lewinsohn, Mathilde Nguyen, Lourdes Velo-Suarez, Anthony Herzig, Thomas E Ludwig, Helen Castillo-Madeen, Donald F Conrad, Emmanuelle Génin, Gaëlle Marenne","doi":"10.1159/000543671","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Next-generation sequencing (NGS) data analysis has become an integral part of clinical genetic diagnosis, raising the question of variant prioritization. The Population Sampling Probability (PSAP) method has been developed to tackle the issue of variant prioritization in the exome of a single patient, by leveraging allele frequencies from population databases and a variant pathogenicity score.</p><p><strong>Methods: </strong>Here, we present Easy-PSAP, a completely new implementation of the PSAP method comprising two user-friendly and highly adaptable pipelines. Easy-PSAP allows the gene-based recalibration of any in silico pathogenicity prediction score compared to scores of variants seen in the general population, including popular scores like CADD or AlphaMissense. Easy-PSAP can evaluate genetic variants at the scale of a whole exome or a whole genome using information from the latest population and annotation databases.</p><p><strong>Results: </strong>Through simulations on synthetic disease exomes, we show that Easy-PSAP is able to rank more than 50% of causal pathogenic variants in the top 10 variants for an autosomal dominant model of transmission and in top 1 for an autosomal recessive model of transmission.</p><p><strong>Discussion: </strong>These findings, along with the accessibility of the pipeline to both researchers and clinicians, make Easy-PSAP a state-of-the-art tool for variant prioritization in NGS data that can continue to evolve as new frameworks and databases become available. Easy-PSAP is implemented in R and bash within an open-source Snakemake framework. It is available on GitHub alongside conda environments containing the required dependencies (<ext-link ext-link-type=\"uri\" xlink:href=\"https://github.com/msogloblinsky/Easy-PSAP\" xmlns:xlink=\"http://www.w3.org/1999/xlink\">https://github.com/msogloblinsky/Easy-PSAP</ext-link>).</p>","PeriodicalId":13226,"journal":{"name":"Human Heredity","volume":" ","pages":"33-40"},"PeriodicalIF":1.5000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12245139/pdf/","citationCount":"0","resultStr":"{\"title\":\"Easy-PSAP: An Integrated Workflow to Prioritize Pathogenic Variants in Sequence Data from a Single Individual.\",\"authors\":\"Marie-Sophie C Ogloblinsky, Marc B Gros-La-Faige, Daniel P Lewinsohn, Mathilde Nguyen, Lourdes Velo-Suarez, Anthony Herzig, Thomas E Ludwig, Helen Castillo-Madeen, Donald F Conrad, Emmanuelle Génin, Gaëlle Marenne\",\"doi\":\"10.1159/000543671\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Next-generation sequencing (NGS) data analysis has become an integral part of clinical genetic diagnosis, raising the question of variant prioritization. The Population Sampling Probability (PSAP) method has been developed to tackle the issue of variant prioritization in the exome of a single patient, by leveraging allele frequencies from population databases and a variant pathogenicity score.</p><p><strong>Methods: </strong>Here, we present Easy-PSAP, a completely new implementation of the PSAP method comprising two user-friendly and highly adaptable pipelines. Easy-PSAP allows the gene-based recalibration of any in silico pathogenicity prediction score compared to scores of variants seen in the general population, including popular scores like CADD or AlphaMissense. Easy-PSAP can evaluate genetic variants at the scale of a whole exome or a whole genome using information from the latest population and annotation databases.</p><p><strong>Results: </strong>Through simulations on synthetic disease exomes, we show that Easy-PSAP is able to rank more than 50% of causal pathogenic variants in the top 10 variants for an autosomal dominant model of transmission and in top 1 for an autosomal recessive model of transmission.</p><p><strong>Discussion: </strong>These findings, along with the accessibility of the pipeline to both researchers and clinicians, make Easy-PSAP a state-of-the-art tool for variant prioritization in NGS data that can continue to evolve as new frameworks and databases become available. Easy-PSAP is implemented in R and bash within an open-source Snakemake framework. It is available on GitHub alongside conda environments containing the required dependencies (<ext-link ext-link-type=\\\"uri\\\" xlink:href=\\\"https://github.com/msogloblinsky/Easy-PSAP\\\" xmlns:xlink=\\\"http://www.w3.org/1999/xlink\\\">https://github.com/msogloblinsky/Easy-PSAP</ext-link>).</p>\",\"PeriodicalId\":13226,\"journal\":{\"name\":\"Human Heredity\",\"volume\":\" \",\"pages\":\"33-40\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12245139/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Heredity\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1159/000543671\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/6/10 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Heredity","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1159/000543671","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/10 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Easy-PSAP: An Integrated Workflow to Prioritize Pathogenic Variants in Sequence Data from a Single Individual.
Introduction: Next-generation sequencing (NGS) data analysis has become an integral part of clinical genetic diagnosis, raising the question of variant prioritization. The Population Sampling Probability (PSAP) method has been developed to tackle the issue of variant prioritization in the exome of a single patient, by leveraging allele frequencies from population databases and a variant pathogenicity score.
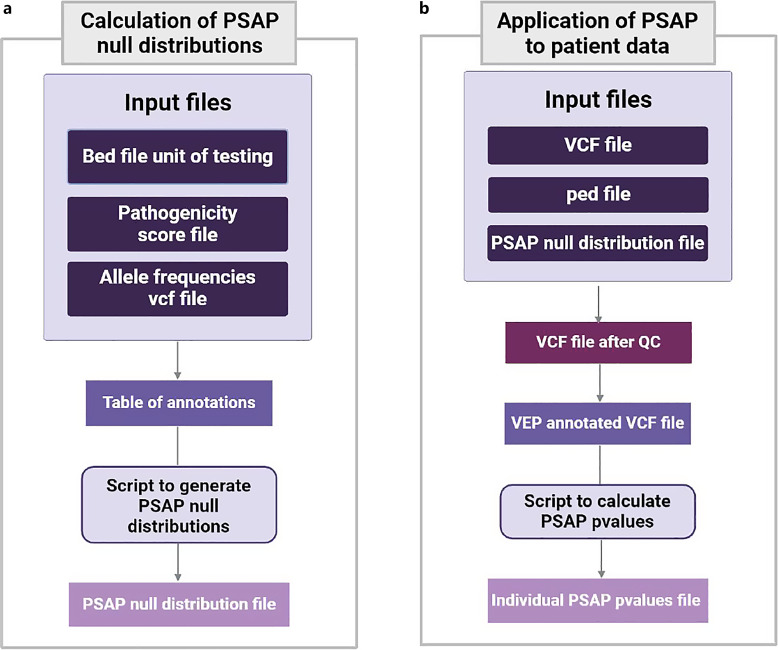
Methods: Here, we present Easy-PSAP, a completely new implementation of the PSAP method comprising two user-friendly and highly adaptable pipelines. Easy-PSAP allows the gene-based recalibration of any in silico pathogenicity prediction score compared to scores of variants seen in the general population, including popular scores like CADD or AlphaMissense. Easy-PSAP can evaluate genetic variants at the scale of a whole exome or a whole genome using information from the latest population and annotation databases.
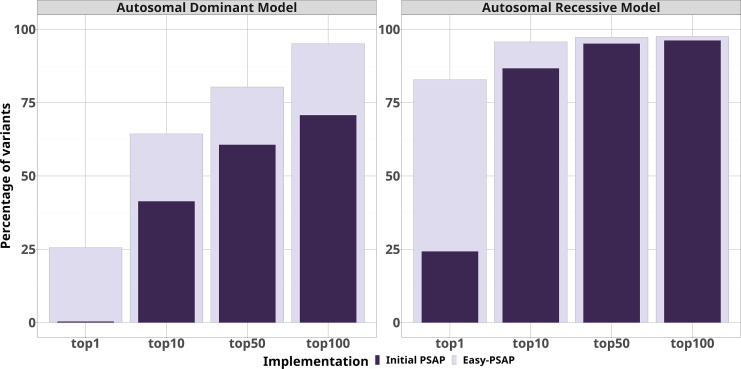
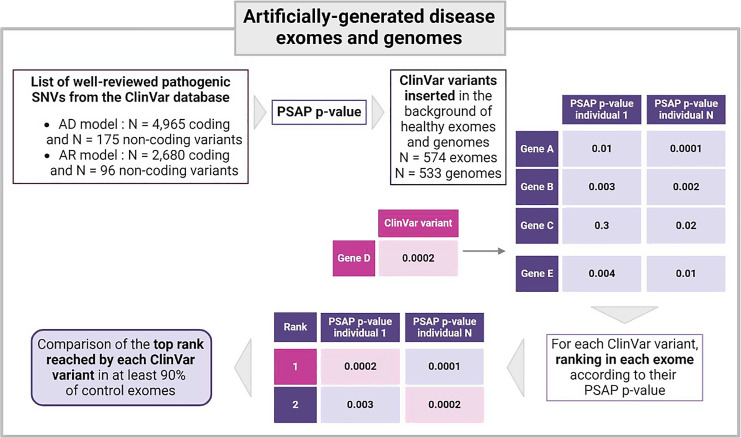
Results: Through simulations on synthetic disease exomes, we show that Easy-PSAP is able to rank more than 50% of causal pathogenic variants in the top 10 variants for an autosomal dominant model of transmission and in top 1 for an autosomal recessive model of transmission.
Discussion: These findings, along with the accessibility of the pipeline to both researchers and clinicians, make Easy-PSAP a state-of-the-art tool for variant prioritization in NGS data that can continue to evolve as new frameworks and databases become available. Easy-PSAP is implemented in R and bash within an open-source Snakemake framework. It is available on GitHub alongside conda environments containing the required dependencies (https://github.com/msogloblinsky/Easy-PSAP).
期刊介绍:
Gathering original research reports and short communications from all over the world, ''Human Heredity'' is devoted to methodological and applied research on the genetics of human populations, association and linkage analysis, genetic mechanisms of disease, and new methods for statistical genetics, for example, analysis of rare variants and results from next generation sequencing. The value of this information to many branches of medicine is shown by the number of citations the journal receives in fields ranging from immunology and hematology to epidemiology and public health planning, and the fact that at least 50% of all ''Human Heredity'' papers are still cited more than 8 years after publication (according to ISI Journal Citation Reports). Special issues on methodological topics (such as ‘Consanguinity and Genomics’ in 2014; ‘Analyzing Rare Variants in Complex Diseases’ in 2012) or reviews of advances in particular fields (‘Genetic Diversity in European Populations: Evolutionary Evidence and Medical Implications’ in 2014; ‘Genes and the Environment in Obesity’ in 2013) are published every year. Renowned experts in the field are invited to contribute to these special issues.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


