Renana Schwartz, Amit Hadar-Volk, Kwangho Nam and Dan T. Major*,
{"title":"基于模板的酶dock最大共同子结构自动鉴定对接:机械与抑制剂对接","authors":"Renana Schwartz, Amit Hadar-Volk, Kwangho Nam and Dan T. Major*, ","doi":"10.1021/acs.jcim.5c0020110.1021/acs.jcim.5c00201","DOIUrl":null,"url":null,"abstract":"<p >EnzyDock is a multistate, multiscale CHARMM-based docking program which enables mechanistic docking, i.e., modeling enzyme reactions by docking multiple reaction states, like substrates, intermediates, transition states, and products to the enzyme, in addition to standard protein–ligand docking. To achieve docking of multiple reaction states with similar poses (i.e., consensus docking), EnzyDock employs consensus pose restraints of the docked ligand states relative to a docking template. In the current work, we present an implementation of a Maximum Common Substructure (MCS)-guided docking strategy using EnzyDock, enabling the automatic detection of similarity among query ligands. Specifically, the MCS multistate approach is employed to efficiently dock ligands along enzyme reaction coordinates, including reactants, intermediates, and products, which allows efficient and robust mechanistic docking. To demonstrate the effectiveness of the MCS strategy in modeling enzymes, it is first applied to two highly complex enzyme reaction cascades catalyzed by the diterpene synthase CotB2 and the Diels–Alderase LepI. In addition, the MCS strategy is applied to dock enzyme inhibitors using cocrystallized inhibitors or substrates to guide the docking in the enzymes dihydrofolate reductase and the SARS-CoV-2 enzyme M<sup>pro</sup>. The latter case exemplifies the use of MCS with EnzyDock’s covalent docking capabilities and QM/MM scoring option. We show that different protocols of the implemented MCS algorithm are needed to obtain mechanistic consistency (i.e., similar poses) in mechanistic docking or to accurately dock chemically diverse ligands in inhibitor docking. Although the current implementation is specific for EnzyDock, the findings should be general and transferable to additional docking programs.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 11","pages":"5596–5611 5596–5611"},"PeriodicalIF":5.3000,"publicationDate":"2025-05-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Template-Based Docking Using Automated Maximum Common Substructure Identification with EnzyDock: Mechanistic and Inhibitor Docking\",\"authors\":\"Renana Schwartz, Amit Hadar-Volk, Kwangho Nam and Dan T. Major*, \",\"doi\":\"10.1021/acs.jcim.5c0020110.1021/acs.jcim.5c00201\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >EnzyDock is a multistate, multiscale CHARMM-based docking program which enables mechanistic docking, i.e., modeling enzyme reactions by docking multiple reaction states, like substrates, intermediates, transition states, and products to the enzyme, in addition to standard protein–ligand docking. To achieve docking of multiple reaction states with similar poses (i.e., consensus docking), EnzyDock employs consensus pose restraints of the docked ligand states relative to a docking template. In the current work, we present an implementation of a Maximum Common Substructure (MCS)-guided docking strategy using EnzyDock, enabling the automatic detection of similarity among query ligands. Specifically, the MCS multistate approach is employed to efficiently dock ligands along enzyme reaction coordinates, including reactants, intermediates, and products, which allows efficient and robust mechanistic docking. To demonstrate the effectiveness of the MCS strategy in modeling enzymes, it is first applied to two highly complex enzyme reaction cascades catalyzed by the diterpene synthase CotB2 and the Diels–Alderase LepI. In addition, the MCS strategy is applied to dock enzyme inhibitors using cocrystallized inhibitors or substrates to guide the docking in the enzymes dihydrofolate reductase and the SARS-CoV-2 enzyme M<sup>pro</sup>. The latter case exemplifies the use of MCS with EnzyDock’s covalent docking capabilities and QM/MM scoring option. We show that different protocols of the implemented MCS algorithm are needed to obtain mechanistic consistency (i.e., similar poses) in mechanistic docking or to accurately dock chemically diverse ligands in inhibitor docking. Although the current implementation is specific for EnzyDock, the findings should be general and transferable to additional docking programs.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 11\",\"pages\":\"5596–5611 5596–5611\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-05-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00201\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00201","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
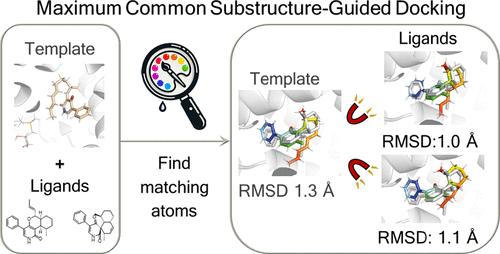
Template-Based Docking Using Automated Maximum Common Substructure Identification with EnzyDock: Mechanistic and Inhibitor Docking
EnzyDock is a multistate, multiscale CHARMM-based docking program which enables mechanistic docking, i.e., modeling enzyme reactions by docking multiple reaction states, like substrates, intermediates, transition states, and products to the enzyme, in addition to standard protein–ligand docking. To achieve docking of multiple reaction states with similar poses (i.e., consensus docking), EnzyDock employs consensus pose restraints of the docked ligand states relative to a docking template. In the current work, we present an implementation of a Maximum Common Substructure (MCS)-guided docking strategy using EnzyDock, enabling the automatic detection of similarity among query ligands. Specifically, the MCS multistate approach is employed to efficiently dock ligands along enzyme reaction coordinates, including reactants, intermediates, and products, which allows efficient and robust mechanistic docking. To demonstrate the effectiveness of the MCS strategy in modeling enzymes, it is first applied to two highly complex enzyme reaction cascades catalyzed by the diterpene synthase CotB2 and the Diels–Alderase LepI. In addition, the MCS strategy is applied to dock enzyme inhibitors using cocrystallized inhibitors or substrates to guide the docking in the enzymes dihydrofolate reductase and the SARS-CoV-2 enzyme Mpro. The latter case exemplifies the use of MCS with EnzyDock’s covalent docking capabilities and QM/MM scoring option. We show that different protocols of the implemented MCS algorithm are needed to obtain mechanistic consistency (i.e., similar poses) in mechanistic docking or to accurately dock chemically diverse ligands in inhibitor docking. Although the current implementation is specific for EnzyDock, the findings should be general and transferable to additional docking programs.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


