评价mfi -沸石相变预测的模拟精度
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们研究了三种流行的沸石经典模型的适用性,其中包括使用分子动力学模拟再现硅质MFI沸石中温度依赖的正交-单斜相变的灵活性的影响。在这些条件下,有关结构变化的工作和研究是有限的;因此,我们利用计算x射线衍射(XRD)图和红外光谱在等温-等压系综条件下研究了这一现象。测试的力场都不能正确地再现相变。我们发现,使用Demontis等人的力场,结构会崩溃,而Nicholas等人以及Hill和Sauer等人的力场被证明过于灵活,会根据MFI的初始阶段产生新的稳定状态。以这些平衡为新基线,进行了进一步的模拟,以确定是否有可能发生进一步的相变。使用XRD和几何准则,我们引入了量化参考相的发散,我们确实发现了转变,但同样依赖于初始相的对称性。研究发现,最初处于更对称的正交态的系统不会从这些构型转变,而那些最初处于单斜态的系统会,但会转变到全新的相。本文章由计算机程序翻译,如有差异,请以英文原文为准。
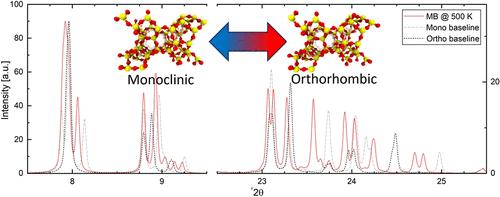
Evaluating Simulation Accuracy for the Prediction of MFI-Zeolite Phase Transitions
We studied the applicability of three popular classical models for zeolites, which include the effects of flexibility in their ability to reproduce the temperature-dependent orthorhombic-monoclinic phase transition in silicious MFI zeolites using molecular dynamics simulations. Work and research on structural changes under these conditions are limited; therefore, we investigated this under isothermal–isobaric ensemble conditions with adjustable cell lengths using computational X-ray diffraction (XRD) patterns and infrared spectra. None of the tested force fields were able to correctly reproduce the phase changes. We found that the structure collapsed using the force field by Demontis et al., while the force fields by Nicholas et al. and Hill and Sauer proved too flexible, creating new stable states dependent on the initial phase of MFI. With these equilibria as new baselines, further simulations were performed to find if any further phase changes were possible. Using XRD and a geometric criterion we introduced to quantify divergences from reference phases, we did find transitions, but again with dependencies on the initial phase’s symmetry. Systems initially in more symmetric orthorhombic states were found to not transition away from those configurations, while those in initially monoclinic states did, but did so to completely new phases.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


