{"title":"MORE-RNAseq:基于RNA-seq数据定量逆转录LINE1表达的管道。","authors":"Yutaka Nakachi, Jianbin Du, Risa Watanabe, Yutaro Yanagida, Miki Bundo, Kazuya Iwamoto","doi":"10.3389/fbinf.2025.1575346","DOIUrl":null,"url":null,"abstract":"<p><p>Retrotransposon long interspersed nuclear element-1 (LINE-1, L1) constitutes a large proportion of the mammalian genome. A fraction of L1s, which have no deleterious mutations in the structure, can amplify their copies via a process called retrotransposition (RT). RT affects genome stability and gene expression and is involved in the pathogenesis of many hereditary diseases. Measuring expression of RT-capable L1s (rc-L1s) among the hundreds of thousands of non rc-L1s is an essential step to understand the impact of RT. We developed mobile element-originated read enrichment from RNA-seq data (MORE-RNAseq), a pipeline for calculating expression of rc-L1s using manually curated L1 references in humans and mice. MORE-RNAseq allows for quantification of expression levels of overall (sum of the expression of all rc-L1s) and individual rc-L1s with consideration of the genomic context. We applied MORE-RNAseq to publicly available RNA-seq data of human and mouse cancer cell lines from the studies that reported increased L1 expression. We found the significant increase of rc-L1 expressions at the overall level in both inter- and intragenic contexts. We also identified differentially expressed rc-L1s at the locus level, which will be the important candidates for downstream analysis. We also applied our method to young and aged human muscle RNA-seq data with no prior information about L1 expression, and found a significant increase of rc-L1 expression in the aged samples. Our method will contribute to understand the role of rc-L1s in various physiological and pathophysiological conditions using standard RNA-seq data. All scripts are available at https://github.com/molbrain/MORE-RNAseq.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"5 ","pages":"1575346"},"PeriodicalIF":3.9000,"publicationDate":"2025-05-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12138260/pdf/","citationCount":"0","resultStr":"{\"title\":\"MORE-RNAseq: a pipeline for quantifying retrotransposition-capable LINE1 expression based on RNA-seq data.\",\"authors\":\"Yutaka Nakachi, Jianbin Du, Risa Watanabe, Yutaro Yanagida, Miki Bundo, Kazuya Iwamoto\",\"doi\":\"10.3389/fbinf.2025.1575346\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Retrotransposon long interspersed nuclear element-1 (LINE-1, L1) constitutes a large proportion of the mammalian genome. A fraction of L1s, which have no deleterious mutations in the structure, can amplify their copies via a process called retrotransposition (RT). RT affects genome stability and gene expression and is involved in the pathogenesis of many hereditary diseases. Measuring expression of RT-capable L1s (rc-L1s) among the hundreds of thousands of non rc-L1s is an essential step to understand the impact of RT. We developed mobile element-originated read enrichment from RNA-seq data (MORE-RNAseq), a pipeline for calculating expression of rc-L1s using manually curated L1 references in humans and mice. MORE-RNAseq allows for quantification of expression levels of overall (sum of the expression of all rc-L1s) and individual rc-L1s with consideration of the genomic context. We applied MORE-RNAseq to publicly available RNA-seq data of human and mouse cancer cell lines from the studies that reported increased L1 expression. We found the significant increase of rc-L1 expressions at the overall level in both inter- and intragenic contexts. We also identified differentially expressed rc-L1s at the locus level, which will be the important candidates for downstream analysis. We also applied our method to young and aged human muscle RNA-seq data with no prior information about L1 expression, and found a significant increase of rc-L1 expression in the aged samples. Our method will contribute to understand the role of rc-L1s in various physiological and pathophysiological conditions using standard RNA-seq data. All scripts are available at https://github.com/molbrain/MORE-RNAseq.</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"5 \",\"pages\":\"1575346\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2025-05-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12138260/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2025.1575346\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2025.1575346","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
MORE-RNAseq: a pipeline for quantifying retrotransposition-capable LINE1 expression based on RNA-seq data.
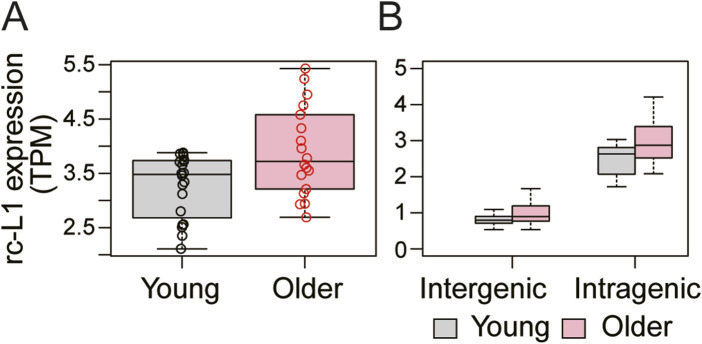
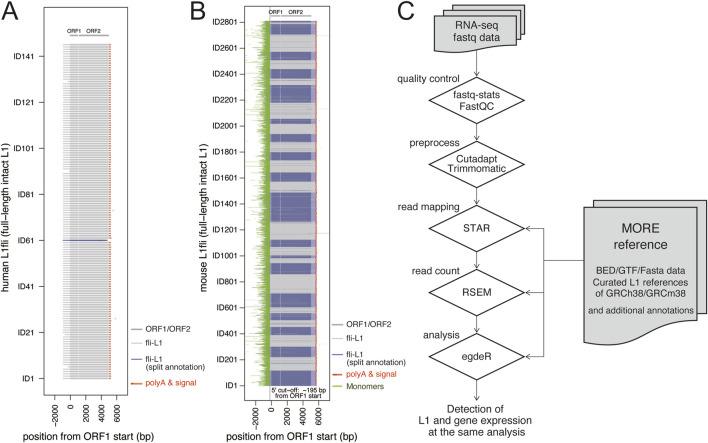
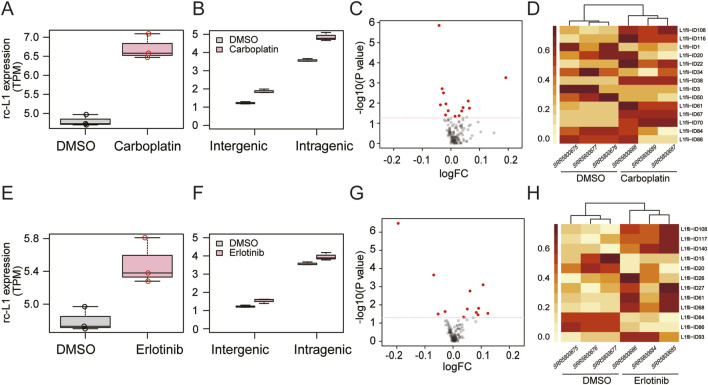
Retrotransposon long interspersed nuclear element-1 (LINE-1, L1) constitutes a large proportion of the mammalian genome. A fraction of L1s, which have no deleterious mutations in the structure, can amplify their copies via a process called retrotransposition (RT). RT affects genome stability and gene expression and is involved in the pathogenesis of many hereditary diseases. Measuring expression of RT-capable L1s (rc-L1s) among the hundreds of thousands of non rc-L1s is an essential step to understand the impact of RT. We developed mobile element-originated read enrichment from RNA-seq data (MORE-RNAseq), a pipeline for calculating expression of rc-L1s using manually curated L1 references in humans and mice. MORE-RNAseq allows for quantification of expression levels of overall (sum of the expression of all rc-L1s) and individual rc-L1s with consideration of the genomic context. We applied MORE-RNAseq to publicly available RNA-seq data of human and mouse cancer cell lines from the studies that reported increased L1 expression. We found the significant increase of rc-L1 expressions at the overall level in both inter- and intragenic contexts. We also identified differentially expressed rc-L1s at the locus level, which will be the important candidates for downstream analysis. We also applied our method to young and aged human muscle RNA-seq data with no prior information about L1 expression, and found a significant increase of rc-L1 expression in the aged samples. Our method will contribute to understand the role of rc-L1s in various physiological and pathophysiological conditions using standard RNA-seq data. All scripts are available at https://github.com/molbrain/MORE-RNAseq.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


